1. 抗HIV-1宿主防御因子としてのAPOBEC3タンパク質
ヒト免疫不全ウイルスI型(human immunodeficiency virus type 1:HIV-1)はレトロウイルス科レンチウイルス属の一本鎖(プラス鎖)のRNAウイルスであり,後天性免疫不全症候群(acquired immunodeficiency syndrome:AIDS)の原因ウイルスとして知られている.1983年にLuc MontagnierとFrançoise Barré-SinoussiによってAIDSの原因病原体として分離・同定1)されて以来,現在までのHIV-1感染症による累計感染者数は7000万人以上,2013年末における全世界のHIV-1感染者数は3500万人と推定されている(http://www.unaids.org/globalreport/).
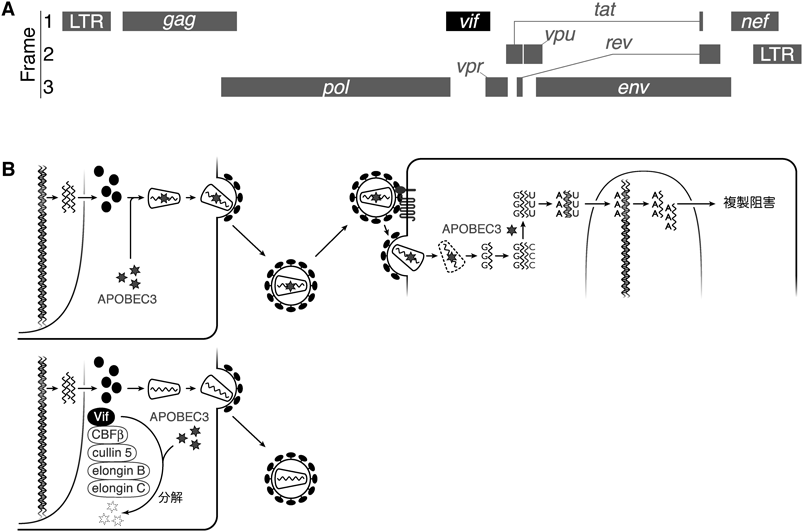
HIV-1のゲノムサイズは約10 kb程度であり,九つの遺伝子をコードしている(図1A).また,HIV-1はその複製過程において,さまざまなヒトの(細胞)タンパク質を利用することが知られている2).一方,ヒトゲノムは,HIV-1の複製を阻害する活性を有するタンパク質(「宿主防御因子(restriction factor)」,あるいは「内因性免疫(intrinsic immunity)」と呼ばれる)をコードしていることが明らかになってきている3).その代表例として,本特集のトピックであるAPOBEC3タンパク質が知られている.
ヒトAPOBEC3ファミリーは,七つのタンパク質(APOBEC3A, B, C, D, F, G, H)から構成される(詳細は「緒言」図1を参照とする).これらのタンパク質のうち,HIV-1の主たる感染細胞であるヒトCD4陽性T細胞に内在的に発現するAPOBEC3FとAPOBEC3Gが特に強力な抗HIV-1効果を発揮することが,筆者らの研究を含めたこれまでの研究で明らかとなっている(なお,APOBEC3Hも抗HIV-1活性を有していることが知られているが,これについては本稿後半で詳述する)4, 5).
HIV-1感染CD4陽性T細胞に内在的に発現するAPOBEC3タンパク質は,ウイルス産生の際にウイルス粒子に取り込まれ,新規の感染細胞へと持ち込まれる.そして,その細胞に持ち込まれたAPOBEC3タンパク質は,逆転写反応の過程で生成されるマイナス鎖DNAの中のシトシン(cytosine:C)のアミノ基(NH2基)を,酵素反応により脱アミノ化する(図1B上段).この脱アミノ化反応によってCはウラシル(uracil:U)へと変換される(C-to-U変異).HIV-1のマイナス鎖DNA中のC-to-U変異は,プラス鎖のグアニン(guanine:G)がアデニン(adenine:A)へと変換されるG-to-A変異として帰結する.このようなAPOBEC3タンパク質の酵素反応によって,HIV-1ゲノム中にG-to-A変異が蓄積され,それらがナンセンス変異あるいはミスセンス変異となり,HIV-1の複製能・感染能を失効させる(図1B).
2. アクセサリータンパク質Vif:宿主防御因子APOBEC3のアンタゴニスト
九つのHIV-1遺伝子のうち,五つは構造・機能・調節タンパク質(Gag, Pol, Env, Tat, Rev)をコードしており,これらはすべてウイルス複製に必須である.これらに加え,HIV-1は,viral infectivity factor(Vif),viral protein U(Vpu),viral protein R(Vpr),negative factor(Nef)という四つのタンパク質をコードしており,これらは総じて“アクセサリータンパク質”と呼ばれる(図1A)6).これまでの研究から,Vifは,Cullin 5, Elongin B/C, CBF-βなどの細胞由来のE3ユビキチンリガーゼ複合体を動員し,APOBEC3タンパク質をユビキチン/プロテアソーム経路依存的に分解することによって,APOBEC3タンパク質による抗HIV-1活性を拮抗阻害することが明らかとなっている7).なお,本稿では詳細にはふれないが,たとえばVpuは別の抗HIV-1宿主防御因子tetherinを拮抗することなどがわかっている8).つまり,Vifを含めたHIV-1のアクセサリータンパク質は,ウイルスの進化の過程において,ウイルス複製をより効率よく遂行するために必要に応じて獲得されてきたものと推察されている.なお,VifとAPOBEC3の構造生物学的な理解については岩谷らの稿「2. APOBEC3とVifの構造からみえてきたウイルス戦略」を参照されたい.
3. “ヒト化マウス”:HIV-1感染病態を再現する小動物モデルを用いてのHIV-1とAPOBEC3の相克
1)ヒト化マウスモデルの作出9, 10)
上述のように,AIDSを含めたHIV-1感染症は,原因病原体の分離・同定から30年以上経った現在においても,多数の新規感染者を世界中で生み出し続ける喫緊の感染症である.1990年代後半にさまざまな作用機序の抗HIV-1薬が多数開発され,それによって多剤併用療法が導入され,HIV-1感染症に対する治療成績は劇的に向上した.しかしながら,HIV-1を生体から完全に排除(eradication)するための根治療法はいまだに確立されていない.その一因として,このウイルスの標的細胞がCD4陽性T細胞という比較的寿命の長い細胞であること,そして,HIV-1の宿主域がヒトに限られているために,HIV-1の感染病態を再現できる動物モデルが存在しなかったことがあげられる.
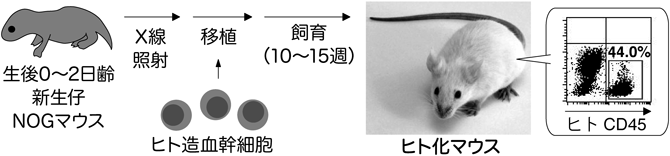
HIV-1感染病態を再現できる新たな動物モデルを作製するために,筆者らは,重度複合免疫不全マウスであるNOD/SCID/Il2rg−/−マウス(NOGマウス;実験動物中央研究所が作製11))にヒトCD34陽性造血幹細胞を移植することにより,ヒト造血能を1年以上維持できる「ヒト化マウス」を作出した(図2).作出したヒト化マウスは,HIV-1の増殖を30週以上維持することが可能であり,また,末梢血中のCD4陽性T細胞の漸進的減少に代表されるHIV-1感染病態を忠実に再現する12, 13).
これまで,株化細胞や初代培養細胞を用いたin vitroにおける詳細な実験解析から,HIV-1複製過程におけるウイルスとヒトタンパク質との相互作用を,「ウイルスタンパク質と宿主防御因子の相互作用(せめぎ合い)」として理解することが可能となってきた.一方,臨床検体などを用いた研究により,HIV-1の病態発現原理についての知見も増えつつある.しかしながら,HIV-1感染を持続的に再現できる適切な動物モデルがなかったため,培養細胞を用いたミクロな知見と,臨床から得られたマクロな知見をつなぐこと,すなわち,HIV-1アクセサリータンパク質と宿主防御因子の生体内における相互作用,およびそれぞれの機能については不明であった.筆者らは,リバースジェネティクス法を駆使して作製したさまざまなアクセサリー遺伝子欠損・変異体ウイルスとヒト化マウスモデルを用い,HIV-1感染病態におけるアクセサリータンパク質と宿主防御因子の役割を明らかにしてきた.以下,これまでに得られている筆者らの知見の中で,特にAPOBEC3タンパク質とVifの相克・せめぎ合いについて得られた最近の知見を紹介する.
2)vif欠損HIV-1感染ヒト化マウス14)
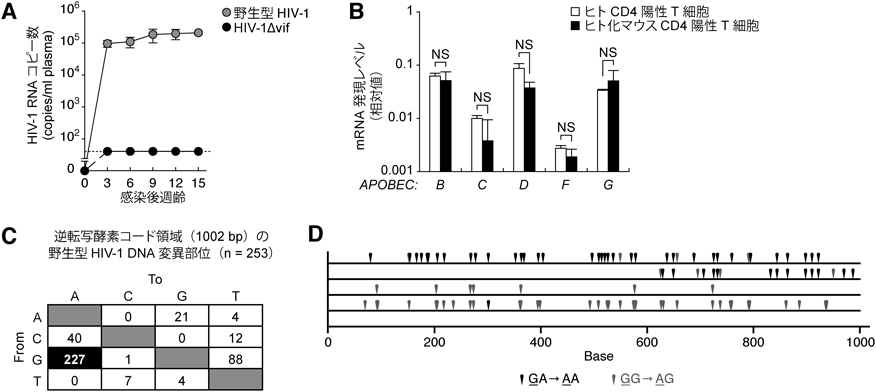
生体内HIV-1感染ダイナミクスにおけるVifとAPOBEC3タンパク質それぞれの機能を明らかにすることを目的として,筆者らはまず,リバースジェネティクス法によってvif欠損HIV-1(HIV-1Δvif)を作製した.HIV-1Δvifと野生型HIV-1をそれぞれヒト化マウスに接種した結果,野生型HIV-1は効率よく増殖したのに対し,HIV-1Δvifはヒト化マウス内でまったく増殖しなかった(図3A).また,ヒト健常人末梢血とヒト化マウス脾臓それぞれのCD4陽性T細胞におけるAPOBEC3遺伝子(APOBEC3B, C, D, F, G)の発現レベルをリアルタイムRT-PCRにより定量した結果,測定したすべての遺伝子の発現レベルは,ヒトとヒト化マウスで同等であった(図3B).このことから,ヒト化マウス内のヒトCD4陽性T細胞での内在性APOBEC3タンパク質の発現環境は,ヒトのそれを忠実に反映したものであることが確認された.
次に,内在性APOBEC3タンパク質による抗ウイルス効果を評価するために,感染後15週齢の感染マウスの脾臓からDNAを抽出し,HIV-1のプロウイルス(ヒトゲノムに組み込まれたウイルスDNA)の配列を解析した.その結果,顕著なG-to-A変異が確認されたことから(図3C),ヒトCD4陽性T細胞に内在的に発現するAPOBEC3タンパク質は,生体内においても高効率にG-to-A変異をウイルス配列に挿入することが示唆された.
上述のように,APOBEC3タンパク質はHIV-1ゲノムにG-to-A変異を挿入するが,それぞれのタンパク質によって指向する隣接塩基配列が異なることが知られている.具体的には,APOBEC3FはGA-to-AA変異(変異するGの次の塩基がA)を,APOBEC3GはGG-to-AG変異(変異するGの次の塩基がG)をそれぞれ好む.配列解析したヒト化マウス内のプロウイルスDNA断片を詳細に検討した結果,あるクローンは顕著なGA-to-AA変異を,一方で別のクローンは顕著なGG-to-AG変異を有していることが確認された(図3D).以上の結果から,生体内のHIV-1増殖においてVifは必須のウイルス因子であること,また,ヒトCD4陽性T細胞に内在的に発現するAPOBEC3タンパク質は,生体内においても強力な抗HIV-1効果を発揮する宿主防御因子であることが明らかとなった.
3)vif変異HIV-1感染ヒト化マウス9)
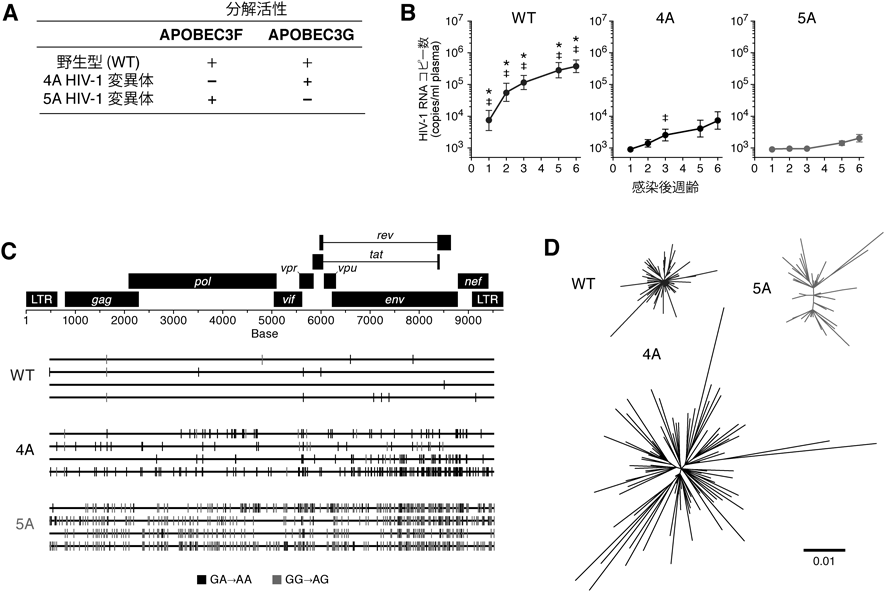
上述の筆者らの研究14)により,APOBEC3タンパク質が生体内HIV-1増殖を強力に抑制することが明らかとなったが,どのAPOBEC3タンパク質,特にAPOBEC3FとAPOBEC3Gのいずれが特にウイルス増殖の抑制に寄与しているかは不明であった.これを明らかにするために,筆者らは,APOBEC3Fのみを分解できない変異体VifをコードするHIV-1(4A変異体)とAPOBEC3Gのみを分解できない変異体VifをコードするHIV-1(5A変異体)をリバースジェネティクス法によってそれぞれ作製し(図4A),これらのウイルスをヒト化マウスに接種した.その結果,4A変異体および5A変異体のヒト化マウスにおける増殖効率は,野生型HIV-1に比して有意に低く,また,5A変異体の増殖効率は,4A変異体のそれに比して有意に低かった(図4B).また,APOBEC3Gは特にGG→AG変異を,APOBEC3FはGA→AA変異をそれぞれ指向することが知られている.そこで,感染後6週の脾臓におけるHIV-1プロウイルスDNAの全長配列を解析した結果,4A変異体HIV-1感染マウスではGA→AA変異が顕著に観察されたのに対し,5A変異体HIV-1感染マウスではGG→AG変異が顕著に観察された(図4C).以上の結果から,生体内HIV-1感染動態において,ヒトCD4陽性T細胞に内在的に発現するAPOBEC3F, APOBEC3Gはともに抗ウイルス活性を示すこと,また,APOBEC3Gの抗ウイルス活性はAPOBEC3Fのそれよりも強力であることが示唆された.さらに興味深いことに,血漿中のウイルスRNA(ヒト化マウスの体内で増殖しているウイルス粒子中のRNA)の配列を単一ウイルスゲノム配列解析した結果,脾臓の細胞内のプロウイルスDNAで観察された変異パターンと異なっており,特に4A変異体HIV-1感染マウスで増殖するウイルスが顕著に多様化していた(図4D).これらの結果は,以下のように解釈できる.APOBEC3GによるGG→AG変異はナンセンス変異を誘導しやすい[たとえば,TGGはトリプトファンをコードするコドンであるが,ここにGG→AG変異が挿入されるとTAG(終止コドン)へと置換される]のに対し,APOBEC3FによるGA→AA変異はナンセンス変異を誘導しにくく,むしろミスセンス変異がウイルスゲノム内に蓄積される.すなわち,APOBEC3Fは強力な抗HIV-1宿主防御因子であると同時に,ウイルスの多様化を促進する機能も有すると示唆される.この事実は,薬剤耐性株や免疫逃避株の出現など,HIV-1にとって有益な進化がAPOBEC3Fによって惹起される可能性を示唆している.実際,筆者らの研究により,感染共受容体をCCR5からCXCR4に変化させたウイルスが4A変異体HIV-1感染マウス特異的に出現すること,そして,その機能変異がAPOBEC3Fによって惹起されたものであることが明らかとなっている9).さらに最近,同様の結果が臨床検体においても確認されることが報告されている10).筆者らの結果は,APOBEC3Fの活性がむしろウイルスに有益に働く可能性,すなわち,宿主にとってもウイルスにとっても諸刃の剣である可能性を示唆している.
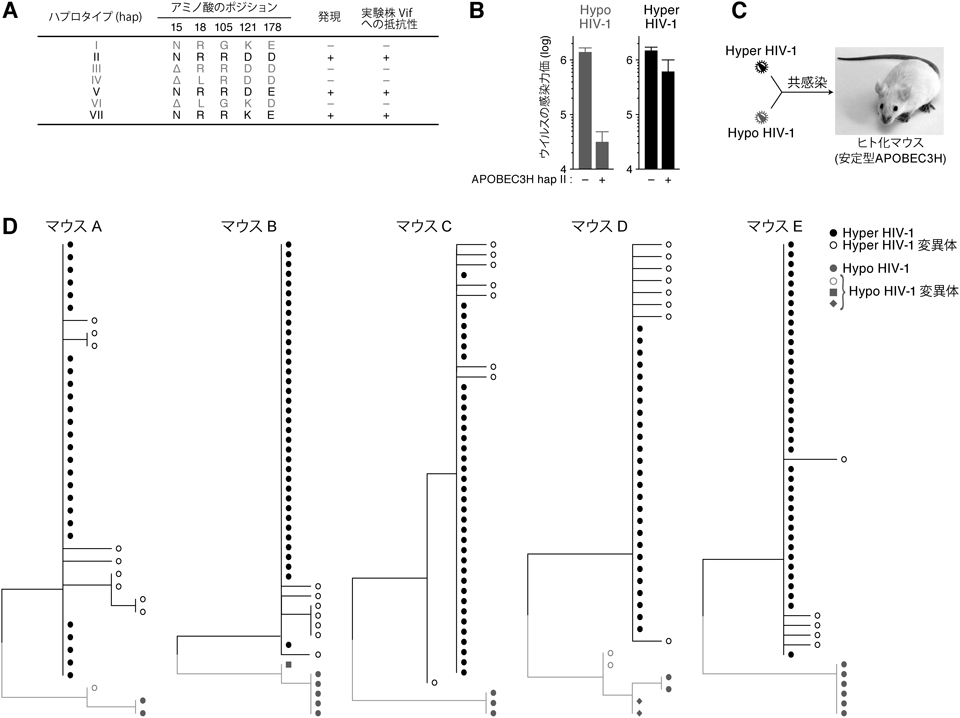
4)APOBEC3Hの遺伝子多型と抗HIV-1効果
上述のように,APOBEC3FとAPOBEC3Gが生体内でのHIV-1増殖・複製に影響を与えることが明らかとなった.これら二つに加え,APOBEC3HもHIV-1複製を抑制する活性を有していることが,これまでの研究から明らかとなっている15–17).興味深いのは,APOBEC3H遺伝子には七つの多型(ハプロタイプ)がある点である(図5A).七つのハプロタイプのうち三つの遺伝子型(ハプロタイプII, V, VII)のAPOBEC3Hは安定的に発現し,抗HIV-1効果を有するのに対し,残る四つの遺伝子型(ハプロタイプI, III, IV, VI)のAPOBEC3Hは,タンパク質として構造的に不安定であるため,ほとんど発現しない15–17).さらに興味深いのは,抗ウイルス活性を示す三つの遺伝子型のAPOBEC3Hは,実験株・臨床分離株を含めたさまざまなVifによる分解に対して抵抗性を示す点である(図5A)15–17).これは,ヒト集団におけるAPOBEC3H遺伝子の多型により,HIV-1の流行・伝播過程において,VifがAPOBEC3H分解活性の獲得と欠失を繰り返しているためと考えられている.なお,APOBEC3H遺伝子の多型は人種によってばらつきがあり,アフリカ系のヒト(ネグロイド)の場合は約50%が安定型APOBEC3H(ハプロタイプII, V, VII)を有しているのに対し,アジア系(モンゴロイド)ではほとんど確認されない18).
最近,安定型APOBEC3Hを分解する活性を有するHIV-1 vif遺伝子が,安定型APOBEC3Hの遺伝子型のHIV-1感染者19),および,培養細胞系を用いたウイルススクリーニング実験18)から同定された.そこで筆者らは,安定型APOBEC3Hを分解できるHIV-1(Hyper HIV-1)と,分解できないHIV-1(Hypo HIV-1)を,それぞれリバースジェネティクス法により作製した(図5B).また,ヒト化マウスのAPOBEC3H遺伝子型をジェノタイピングPCR/シークエンス法により検討し,安定型APOBEC3Hを発現する遺伝子型のヒト化マウスを選別した.その後,安定型APOBEC3H発現ヒト化マウスにHyper HIV-1, Hypo HIV-1を混合・共感染し,どちらのウイルスがより効率よく増殖するかを検討した(図5C).感染6週後における血漿中のウイルスRNA(ヒト化マウスの体内で増殖しているウイルス粒子中のRNA)の配列をRT-PCR/シークエンス法により検討したところ,用いた5匹の安定型APOBEC3Hを発現する遺伝子型のヒト化マウスすべてにおいて,Hyper HIV-1の優位な増殖が確認された(図5D).以上の結果から,ヒトCD4陽性T細胞に内在的に発現する安定型APOBEC3Hもまた,生体内においてHIV-1複製を制御する宿主防御因子であることが実証された(中野,佐藤,小柳ら,論文投稿準備中).