ウイルスは「彷徨える遺伝子」であり,宿主細胞内に侵入してその核酸およびタンパク質合成機構を乗っ取り,感染細胞をウイルス粒子複製の場に変えてしまう.ウイルス感染の標的細胞が中枢神経細胞や肝細胞,あるいは心筋細胞など,宿主の個体維持に決定的に重要な機能を担っている場合,急性感染が短期間に宿主の死につながることがあり,その例は多くの新興感染症に認められる.哺乳動物がウイルスに対して獲得した最も強力な防御反応は,疑いもなく細胞傷害性Tリンパ球を含む獲得免疫機能であり,Tリンパ球に対して抗原提示を行う主要組織適合遺伝子複合体(major histocompatibility complex:MHC)の種を通じた多型が,人類発祥の地からの移動の過程で感受性個体の淘汰により次第に低下していったこと,一方地域別の対立遺伝子数がその地に蔓延するウイルス種の数と相関することが明らかになっている1).一方で,獲得免疫応答によらない細胞レベルの抵抗性機構も複数が知られている2).
レトロウイルスは粒子内に逆転写酵素を持ち,一本鎖RNAからなるウイルスゲノムを二本鎖DNAに変換して,宿主細胞染色体に組込まれる.一般に,レトロウイルスの複製過程は強い細胞傷害効果を伴わず,感染細胞は染色体に組込まれたプロウイルスゲノムを保持したままで分裂・増殖を続け,出芽によりウイルス粒子を産生し続ける.プロウイルス組込みが生殖細胞染色体に生じれば内在性レトロウイルスとなり,子孫個体の体細胞と生殖細胞のすべてがあらかじめ染色体上にレトロウイルス遺伝子を持つ上,ウイルスゲノムの発現と再感染による組込み部位の増加が生じうる.このように,レトロウイルスは真核細胞ゲノムの同一性維持にとって最大の脅威であるため,哺乳類はその進化の過程で複数の対抗機構を獲得してきた.
レトロウイルスゲノムの逆転写過程で生じたマイナス鎖の一本鎖DNAを一次標的とするシチジンデアミナーゼAPOBEC3は,細胞内で機能する最も強力な抗レトロウイルス因子である.細胞レベルで機能するレトロウイルス抵抗性因子には,この他にTRIM5αやSAMHD1があり,マウスの場合は細胞表面でウイルス受容体として利用されるタンパク質(その多くはアミノ酸や無機イオンの輸送体である)の多型や,受容体へのウイルス粒子結合に拮抗するタンパク質,あるいは逆転写産物とタンパク質の複合体であるpreintegration complex(PIC)の細胞内輸送に干渉する因子など(これらはいずれも,内在性レトロウイルスの産物である)が強力な効果を発揮することが知られているが2, 3),進化の過程で最も強い制限効果を発揮してきたのはAPOBEC3であると考えられている4).
APOBEC3によるレトロウイルス複製制限効果は,最初試験管内でのヒト免疫不全ウイルス(HIV)1型(HIV-1)複製における,一件奇妙な「インプリンティング」現象として気づかれた.すなわち,生体から分離されたリンパ球やマクロファージにおけるHIV-1の複製にはアクセサリー遺伝子産物の一つであるVif(viral infectivity factor)が必要であるが,Vif機能を欠損したHIV-1(Δvif)であっても,CD4を発現するHeLa細胞や293T細胞では複製できる.これらの許容性細胞で複製されたΔvifウイルスは,リンパ球やマクロファージなどの非許容性細胞に感染してその染色体に組込まれるが,いったん非許容性細胞から出芽したΔvif由来ウイルスは,非可逆的に複製能が低下しており,非許容性細胞はもとより,許容性細胞でも感染価が著しく低下する5).これら許容性細胞と非許容性細胞の遺伝子発現を比較することで,Vifの標的因子であるAPOBEC3が同定された6).
霊長類レンチウイルスが,それぞれ自然宿主のAPOBEC3に特異的に結合し,その分解を誘導するようなVifを獲得することにより,生体内での複製能を維持していることから,現存の感染性レトロウイルスは,いずれも自然宿主のAPOBEC3に対抗する手段を獲得することで感染能(と病原性)を維持していると考えられた.このため,APOBEC3は現存哺乳類が過去に遭遇してきたレトロウイルスやレトロトランスポゾンに対しては効果を示していたが,現在感染性を示すレンチウイルスや病原性レトロウイルスに対しては,もはや制限効果を示せないとの考え方が一般的であった.その一方で,HIV感染者であるパートナーと非防御的な性行為を継続していながら,数年間にわたりHIV感染が成立しないHIV曝露非感染者で,単球系細胞におけるAPOBEC3Gの発現量が健常者や感染パートナーに比べて有意に高いとの報告があり,直接証明ではないが,ヒトのAPOBEC3Gが,高発現条件下で現在でもHIVに対する制限効果を発揮する可能性が示されていた7).
Vif欠損という条件下で試験管内でのレンチウイルス複製制限効果が示されていたAPOBEC3が,実際生理的にレトロウイルス感染抵抗性に関与していることを最初に示したのは,Susan Rossらであった8).彼女らは,一部の系統のマウスで乳汁を介する垂直感染を繰り返しているベータレトロウイルス,マウス乳がんウイルス(mouse mammary tumor virus:MMTV)を用い,マウスのAPOBEC3が生体内で実際にレトロウイルス複製制限効果を示すことを初めて明らかにした.
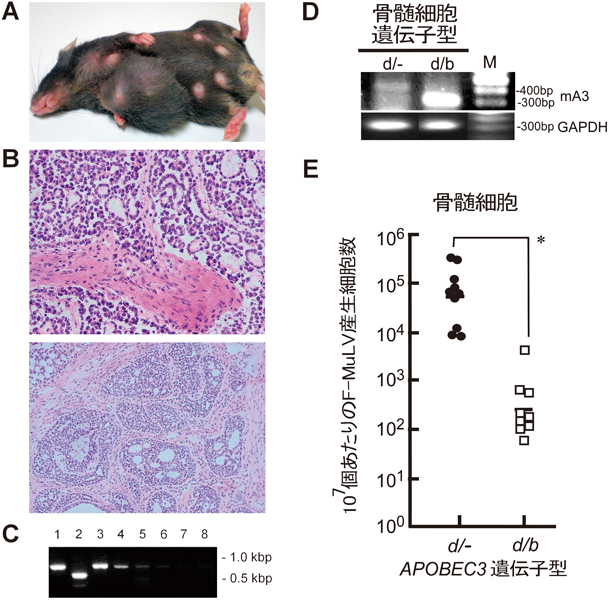
マウス乳がんウイルスは,感染性の内在性MMTVが発現した,あるいは母仔間感染を受けた成熟雌マウスの乳腺上皮で粒子形成が起こり,乳汁を介して新生仔に伝達される.消化管に侵入したMMTVは粘膜リンパ組織の樹状細胞に感染し,ウイルスゲノムのLTR領域にコードされるスーパー抗原によってTリンパ球を活性化することで,全身リンパ組織の標的細胞を増殖させる.MMTVに感染したリンパ球が乳腺内に再循環することで,乳腺発育時および授乳期に活発に分裂する上皮細胞への感染が起こり,細胞がん遺伝子近傍へのプロウイルス組込みががん化につながると同時に,乳汁中へのウイルス粒子出芽を通じて,新生仔への垂直感染が起こる9).Rossらは,マウスAPOBEC3がMMTV粒子に取り込まれ,試験管内でMMTV複製を阻害することを示した8).さらに,APOBEC3遺伝子を標的破壊したマウスを作製し,これに実験的にMMTVを感染させることで,MMTVスーパー抗原によるリンパ球活性化が2倍に高まり,ウイルス複製のレベルも10倍高まることを示して,マウスAPOBEC3がMMTVに対する生理的抵抗因子であることを立証した.さらに,乳腺上皮細胞で発現したAPOBEC3がMMTV粒子に取り込まれ,複製能を低下させることを直接示している10).図1には,我々が観察したAPOBEC3欠損マウスでのMMTV関連乳がん発症例を示す.
マウスAPOBEC3がMMTVに対する生理的抵抗因子であることがRossらにより明らかにされたのとほぼ同じ時期に,日米の少なくとも二つのグループが,まったく別のアプローチからマウス白血病ウイルス(murine leukemia virus:MuLV)に対する抵抗性因子としてのAPOBEC3遺伝子多型を明らかにしつつあった.MuLVは発がんに関与するような宿主由来遺伝子(v-onc)を持たず(オルソレトロウイルス亜科),粒子複製に必要な構造遺伝子だけをゲノムに含む,ガンマレトロウイルス属に分類される感染性の内在性および外来性レトロウイルスであり,自然界(野生マウス)および実験室系のマウスから分離されてきた. AKR系統のマウスでは,マウスおよびラットに感染性を示す内在性の同種指向性レトロウイルスAKVが新生児期から発現して持続感染を起こし,染色体上の内在性多指向性(polytropic)MuLVプロウイルスとの組換えを経て,主にTリンパ球系の白血病・リンパ腫を誘発する. AKVに類似した実験室系のMuLV分離株にモロニー(Moloney:Mo)MuLVがあり,新生仔への接種により,やはり内在性レトロウイルスとの組換え型出現を経てTリンパ球系の白血病を誘発する.一方,北米の野生マウスには野生マウス同種指向性(wild mouse ecotropic:WM-E)MuLVの存在が知られており,水平感染および垂直感染による中枢神経疾患の発症と,リンパ腫・白血病の誘発が報告されている11).
これらAKVやMo-MuLVおよびWM-Eウイルスによる病変発症には,新生仔への感染によるウイルス血症の持続が必要とされ,実際免疫系が完成した成体マウスへの接種では,ウイルスの複製は獲得免疫反応により短期間で終息し,白血病やリンパ腫が起こることはない.これに対して,成体マウスへの接種で短期間に造血系細胞に増殖性病変を誘発し,条件によって致死性病変に至らしめることができるのが,実験室系の分離株であるフレンド(Friend)および類縁のラウシャー(Rauscher)白血病ウイルスである.フレンドウイルス(FV)は,実際には単一のレトロウイルスではなく,複製能を有するガンマレトロウイルスであるフレンドMuLV(F-MuLV)と,複製欠損性であるが急性病原性を有する脾限局巣形成ウイルス(spleen focus-forming virus:SFFV)から構成される複合体である12).F-MuLVは,AKVやMo-MuLVと同様,新生仔への接種で数か月の潜伏期とその間の内在性レトロウイルスとの組換えを経て,赤芽球系または顆粒球・単球系細胞由来の白血病(およびまれにはリンパ腫)を誘発する.しかし,免疫系の完成した成体マウスへの接種では短時日のうちに排除され,病原性を示すことはない.成体マウスにおけるF-MuLVの排除にはCD4陽性Tリンパ球およびBリンパ球が必要であり,T細胞依存性の抗体産生がF-MuLV排除の主要な機構であると考えられる13).一方,F-MuLVとSFFVを複合体として同時接種すると,SFFVのenv遺伝子産物であるgp55が感染赤芽球の急激な増殖を誘発し,成体マウスでも感染から2週間以内に正常重量の10倍を超える脾腫と,多血症が引き起こされる3, 12).さらに条件が整えば,増殖する赤芽球の中からオリゴまたはモノクローナルな赤白血病細胞が出現し,赤芽球の分化停止によりヘマトクリットは低下して,感染個体は著しい肝脾腫により2か月程度で死に至る3).
FV複合体による白血病誘発は,免疫系の完成した成体マウスへの接種で起こるため,多彩な遺伝的背景を持つ純系マウス系統の組合わせと,コンジェニック系統や突然変異マウス,さらにトランスジェニック系統や標的遺伝子ノックアウト系統を活用した,宿主因子の解析が可能となった.実際,FVを用いて複数のレトロウイルス抵抗性宿主遺伝子の存在が記載され,その分子実体が同定されてきた.それらのうち,Fv1遺伝子は前述のPICの細胞内輸送と干渉する内在性レトロウイルス遺伝子産物をコードし14),Fv2遺伝子はSFFVによる赤芽球増殖誘導に関わる造血細胞特異的受容体型チロシンキナーゼStk/Ronの多型をコードする15).また,わが国で発見されたFv4は,細胞表面の同種指向性ウイルス受容体mCAT-1へのF-MuLV吸着と干渉する内在性レトロウイルス遺伝子産物をコードする16).C57BL系のマウス系統,すなわちC57BL/6(B6)やC57BL/10(B10)は,Stk/Ron遺伝子座イントロン10の3塩基欠失により,受容体型チロシンキナーゼの細胞外部分を欠く短いアイソフォームsf-Stkが発現せず,SFFV感染赤芽球の増殖が誘導されないとされてきた.しかし実際には,B6マウスでもCD4陽性Tリンパ球欠損下ではSFFV感染に伴う赤芽球の増殖が(よりゆっくりとではあるが)起こり,多血症を経て赤白血病が生じる13).Bリンパ球単独欠損のB6マウスでは(F-MuLVは排除できないが)SFFVが排除され,初期の脾腫が生じないことから,Fv2遺伝子効果は赤芽球増殖の有無をall-or-none的に決定しているのではなく,B6マウスではSFFV感染赤芽球が,CD4陽性Tリンパ球を介する細胞免疫応答で効率的に排除されていると考えられる.
成体マウスで病原性を示すFVと純系マウスやコンジェニックマウスの特性を活かして,レトロウイルス感染に対する宿主免疫応答を制御する遺伝子群を網羅的に記述し,その分子同定の基礎を築いたのは,Bruce Chesebroである. Chesebroは,ノーベル賞受賞者であるDulbeccoの下でウイルス学を学び,Harvard大学医学部に進学,在学中から免疫グロブリン分子の電子顕微鏡観察で現在も教科書に残る業績をあげた.医師免許取得後,高名なMHC研究者であるH.O. McDevittの下でポストドクを務め,この期間に,FV感染抵抗性はウイルス抗原に対する免疫応答遺伝子現象ではないかとの着想を得た.若くしてモンタナ州Rocky Mountain LaboratoriesでNIAIDのLab. chiefとなったChesebroは,多数のコンジェニックマウス系統を駆使し,Fv1やFv2遺伝子の影響を除いても,なお別の複数の宿主遺伝子が主に免疫応答を介してFV感受性を制御することを明らかにした.
Chesebroが記載したFV抗原に対する免疫応答遺伝子のうち,Rfv1およびRfv2遺伝子はMHC領域にマッピングされ,Rfv1についてはクラスI分子をコードするD遺伝子座の対立遺伝子がその実体であると考えられている17).実際に,抵抗性に関連するDb対立遺伝子産物によって提示されるウイルス遺伝子産物由来のCD8陽性T細胞認識エピトープが同定されており18, 19),このエピトープに反応するTリンパ球の動態も詳しく解析されているが20, 21),これが感染防御に直接関与するか否かは未確定である.Rfv1(=Db)については,Chesebroによる発見の当初からホモ接合体とヘテロ接合体の間で遺伝子量効果(gene dose effect)が知られており,単純な免疫応答遺伝子現象(Db分子による抗原提示の有無)だけではこれを説明できない22).一方,Rfv2遺伝子はクラスIb領域のQ/T遺伝子座の対立遺伝子が実体と考えられるが23),その作用機序は不明である.MHC領域ではその後,クラスIIのAおよびE遺伝子座の対立遺伝子がFV抗原に対するCD4陽性Tリンパ球の応答能を直接制御していることが示され24),こちらはAおよびE遺伝子座の特定の対立遺伝子産物によって提示されるウイルス抗原エピトープが同定されて25, 26),それらを単独で用いることによりFV感染抵抗性が誘導できることが示されている26–28).したがって,免疫応答遺伝子現象としては,クラスII領域遺伝子の効果の方が明確である.
ところが,驚くべきことにMHCの全領域に抵抗性の対立遺伝子を持つH2bハプロタイプのマウスであっても,MHC以外の領域に存在する抵抗性遺伝子を欠くとFVに感受性となってしまう.戻し交配による解析の結果,この非MHC遺伝子は単一の常染色体優性遺伝子であるとされ,Rfv3と命名された29).Rfv3遺伝子の表現型はきわめて興味深く,この遺伝子座に抵抗性の対立遺伝子を持つと考えられるB6やB10背景のマウスでは,FV感染後のウイルス血症が30日以内に消失するが,感受性の対立遺伝子をホモ接合で持つと考えられたBALB/cあるいはA背景のマウスでは,BALB.BやA.BYなどH2bハプロタイプを持っていても,FV感染後30日以上ウイルス血症が持続する.その後,ウイルス血症持続の有無は,ウイルス中和抗体が早期に産生されるか否かで決定されるらしいことが示され30),Rfv3はFV感染に対する抗体産生を制御する,MHCとは異なる遺伝子と考えられるようになった.Rfv3遺伝子の実体解明は困難を極めたが,日米の二つのグループが互いに独立して多数の戻し交配マウスを用いたマッピングを行い31–33),22番染色体にRfv3遺伝子座の存在範囲を絞り込んだ.そして,候補領域遺伝子の発現比較からAPOBEC3遺伝子が最終候補の一つとなり,実際にマウス系統間でAPOBEC3遺伝子に多型が存在することが明らかとなった3).最終的に,両グループがAPOBEC3遺伝子をノックアウトしたB6マウスとその交配子孫の表現型を解析し,B6マウス由来のAPOBEC3対立遺伝子産物が,生体内でFV複製を強く制限する効果を示すことを明らかにした34, 35)(図1).
3. マウスAPOBEC3遺伝子多型の実体と機能差
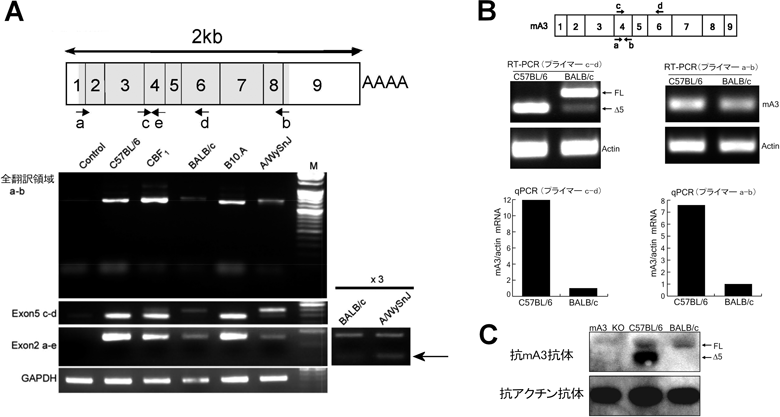
実験室系の純系マウスには,APOBEC3遺伝子座において,B6/B10マウスを代表とする抵抗性系統で発現する対立遺伝子(これを以下mA3bアリルという)と,BALB/cやA系統のマウスで発現する感受性の対立遺伝子(mA3dアリル)の二つの遺伝子型が知られている35, 36).野生マウスに見いだされるAPOBEC3遺伝子型については後述する.マウスAPOBEC3遺伝子座の二つの対立遺伝子の分子実体と,その機能差については,これまでに以下のことが明らかとなっている.
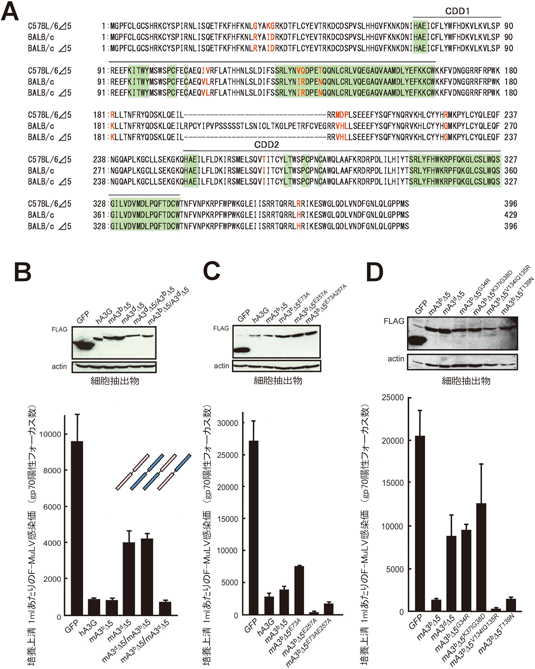
B6/B10マウスで発現するmA3bアリルとBALB/cやA系統マウスで発現するmA3dアリルでは,DNA塩基配列レベルで非常に多くの塩基置換や欠失・挿入があり,その多くはイントロン領域に集積している37).エキソン領域におけるアミノ酸配列多型は,N末端側のZ2ドメインに6か所,C末端側Z3ドメインに6か所の相互置換があり,挿入やフレームシフトはない35).特徴的なことはmA3bとmA3dイントロン2の長さの違いであり36),mA3bアリルではイントロン中に内在性異種指向性(マウスの細胞で発現するが,現存マウスやラットに対する感染性がなく,ウサギや霊長類など,異種の細胞に感染する)レトロウイルスのLTRが組込まれていることである38).
mA3bアリルの存在はMMTVやF-MuLVの複製に対して優性の抑制機能を発揮し,B6マウスからmA3bアリルを欠失させると,生体内でのMMTVの複製がおよそ10倍に8),F-MuLVの複製は100倍に高まって35),病原性が増す(図1).mA3b欠失マウスとmA3dアリルを持つマウスとを交配し,同一の遺伝的背景下でmA3bアリルの効果を解析すると,mA3bアリルはmA3dアリルに対して,生体内で約100倍のF-MuLV複製抑制活性を発揮すると考えられる.これは,以下に示すmRNA発現量の差,エキソン5の有無,タンパク質翻訳効率の差,およびアミノ酸配列多型の影響の複合的な結果であると考えられる.
上記イントロン2への内在性レトロウイルスLTRの組込みは,イントロンエンハンサーとして働くと考えられ,脾細胞で比較するとmA3bアリルからのmRNA発現量は,mA3dアリルからのそれの10倍以上高い35–38)(図2). mRNA発現量の差は,それ自体でレトロウイルス感染抵抗性に影響すると考えられ,実際インターフェロンα処理などによりmA3発現量を増強すると,MMTVに対する複製抑制効果が高まる39).一方,マウスAPOBEC3遺伝子の転写産物には複数のアイソフォームが存在する.全長型は九つのエキソンすべてを含むが,エキソン5欠損型(Δ5)はZ2ドメインとZ3ドメインの間に位置する33アミノ酸残基を欠く(図3).この他に,エキソン2欠損型(Δ2)があり,mA3dの転写産物ではΔ2が多くを占めてこれがドミナントネガティブ型のタンパク質をコードし,APOBEC3機能が低下すると報告されたが34),これは誤りである36).実際には,Δ2の発現量はきわめてわずかで,生理的に意義はないと考えられる(図2).重要なことに,mA3bアリルの転写産物としてはΔ5が大半を占め,一方mA3dアリルの転写産物は全長型が主体で,Δ5はごく少量である(図2).このようにmA3bアリルの転写産物にエキソン5が含まれず,mA3dアリルの転写産物がそれを含む主な理由は,エキソン5内部の一塩基多型(G/C SNP)がこのエキソンのスプライシングの有無に大きな影響を与えるからである37).すなわち,B6由来のmA3bミニゲノムDNAとBALB/c由来mA3dミニゲノムDNAを用いて相互塩基置換を行い,スプライシング産物を比較すると,mA3bの上記エキソンSNPをBALB/c型のCに置換しただけでエキソン5を含んだmRNAが主体となり,同じエキソン内の他の3か所のSNPの相互置換ではスプライシングパターンは変化しない.多数あるイントロン多型もエキソン5のスプライシングにはほとんど影響を与えず,わずかに効果を示すのはイントロン4のスプライシングアクセプター領域に位置する,投げ縄構造形成部位周辺の多型(TCCT反復の有無とT/C SNP)のみである37).
エキソン5の有無は,驚くべきことにマウスAPOBE3タンパク質の翻訳効率に大きな影響を及ぼす.試験管内転写翻訳系を用い,ほぼ等量のmA3bおよびmA3d転写産物を発現させた場合,mA3b由来Δ5タンパク質の翻訳はより急速に起こり,60分後になってもタンパク質発現量はmA3d産物の2倍以上となる.これは,全長型マウスAPOBEC3タンパク質がΔ5に比較して不安定で,より速く分解されるためではなく,翻訳効率が異なるためである.このため,もともとmRNA発現量の高いmA3bアリルを持つマウスの脾臓では,mA3dアリルをホモ接合で持つマウスの脾臓に比べて,APOBEC3タンパク質の発現量が数倍高くなる(図2).なお,同じmA3d由来のΔ5と全長型をそれぞれトランスフェクションしてF-MuLVを感染させ,上清中のウイルスの複製能を比較すると,mA3d由来Δ5は,mA3b産物に比べ弱いながら有意な複製抑制活性を示す35).これは,Δ5の翻訳効率が高いため,粒子あたりにより多くのAPOBEC3分子が取り込まれるためかもしれず,また全長型がウイルス自身のプロテアーゼによりエキソン5内部で切断されるためかもしれない40).しかし,mA3b由来とmA3d由来のΔ5をそれぞれ単独発現させても,mA3bのΔ5の方がF-MuLV複製抑制効果がはるかに高いことから(図3),エキソン5の有無だけでなく,二つの対立遺伝子産物間のアミノ酸配列の違いも,レトロウイルス複製抑制効果に影響することがわかる35).
mA3bのΔ5 cDNAとmA3dのΔ5 cDNAをそれぞれ単独でトランスフェクションした細胞にF-MuLVを感染させ,上清中に産生されたウイルスの二次標的細胞における複製効率を比較すると,10倍以上の差が認められる.この場合,最初に感染させたトランスフェクタントからのウイルス粒子産生数には有意な差はないので35),知られているとおり出芽した粒子中に取り込まれたAPOBEC3が,次に感染した細胞内で複製抑制効果を示すと考えられる.実際,トランスフェクタントから出芽したウイルス粒子中にAPOBEC3の取り込みを検出できる35).そこで,N末端側Z2ドメインの多型とC末端側Z3ドメインの多型のどちらがF-MuLV複製抑制に重要かを解析するため,両対立遺伝子に由来するΔ5型のcDNAクローン間で,Z2コード部分とZ3コード部分を相互に入れ替えたキメラクローンを作製した.これらをトランスフェクションした細胞にF-MuLVを感染させ,上清中に産生されたウイルス粒子の二次標的細胞での複製を比較したところ,N末端側Z2ドメインがmA3b由来である場合に,そうでない場合と比べ明らかに強い複製抑制活性が認められた(図3)35).この場合,キメラ分子間で発現量はほぼ同じであるので,同一タンパク質量が粒子に取り込まれた場合でもアミノ酸配列多型の効果があり,N末端側のアミノ酸配列がmA3b型であると,F-MuLV複製抑制活性が高いことが明らかである.そこで,N末端側Z2ドメインに認められる6個のアミノ酸配列多型について,相互の置換を行ってみた.その結果,驚くべきことにデアミナーゼ活性中心から離れた,N末端寄りの3か所のアミノ酸残基のいずれかをmA3d型へと置換することにより,mA3b産物の示すF-MuLV複製抑制活性が失われることが明らかとなった(図3).これらのアミノ酸残基は,進化の過程で正の選択圧が加わっていることが示されているものである38).
さて,APOBEC3は一本鎖DNAを標的とするシチジンデアミナーゼであり,その抗レトロウイルス効果は逆転写産物におけるシトシン塩基のウラシルへの置換が主な作用機序であると考えられてきた.ところが,mA3各アリルcDNAのトランスフェクタントから出芽したF-MuLVに感染した標的細胞中のプロウイルスを調べてみると,ヒトAPOBEC3Gトランスフェクタントから出芽したF-MuLVの感染で生じたプロウイルスでは,プラス鎖のG-to-A塩基置換が有意に多数検出されたのに対し,mA3b発現細胞に由来するF-MuLVの感染により生じたプロウイルスでは,G-to-A変異の有意な増加は認められなかった35).そこで,マウスAPOBEC3のデアミナーゼ活性中心であるZ2ドメインの73番目のE残基をAに置換した変異体を作製し,これをトランスフェクションした細胞にF-MuLVを感染させて二次標的細胞での複製能を調べてみると,野生型のmA3b由来Δ5を発現させた場合よりやや減弱はしたものの,なお強い複製抑制効果がみられた(図3).このことは,マウスAPOBEC3によるF-MuLVの複製抑制が,少なくとも部分的にデアミナーゼ活性に依存しない機構によっていることを示す.同様に,マウスAPOBEC3によるMMTVの複製抑制もデアミナーゼ活性非依存性であると報告されている39, 41).ところが面白いことに,AKVウイルスに対するマウスAPOBEC3の抑制活性は,デアミナーゼ機能に依存するという42).マウスAPOBEC3が示すデアミナーゼ活性に依存しないF-MuLVおよびMMTV複製抑制活性の分子機構は完全には解明されていないが,MMTVに対しては,その逆転写酵素活性を直接抑制することが示唆されている41).
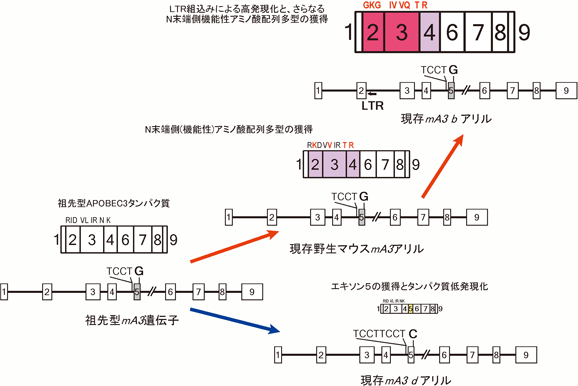
上記の解析結果をまとめると,1)マウスAPOBEC3には遺伝子多型があり,実験室マウスの系統はB6/B10型のmA3bか,BALB/c,A型のmA3dかのいずれかの対立遺伝子を持つ35, 36),2)mA3bはmA3dに比べて生体内でのMMTVおよびF-MuLV複製抑制活性が高く,これらのウイルスに対する生理的な抵抗性因子として機能している8, 34, 35),3)mA3bはmA3dに比べてmRNAレベルでもタンパク質レベルでも発現量が高く,前者の転写活性が高いのは,mA3bのイントロン2に組込まれた内在性レトロウイルスLTRが原因である35–38),4)マウスAPOBEC3の発現量の多寡は,MMTV感染抵抗性と相関する39),5)mA3bから発現するmRNAの大半がエキソン5を欠くのに対し,mA3dの転写産物は全長型が主体であって35, 36),全長型はΔ5に比べて翻訳効率が低い37),6)mA3bの産物とmA3dの産物にはアミノ酸配列多型があり,N末端側のごく少数のアミノ酸残基がmA3bアリル産物の高い抗F-MuLV活性と関連する35),となる.これらのことから最初に予想されるのは,B6/B10マウスの祖先は,a)エキソン5にSNPを獲得することで翻訳効率の高いΔ5を発現するようになり,b)N末端側のアミノ酸置換によりレトロウイルス複製制限活性が高まり,c)イントロン2に異種指向性ウイルスのLTRが内在化することでmRNA転写効率が向上して,最終的にこれら系統の祖先個体が自然界に存在する病原性レトロウイルスに対して高い抵抗性を獲得した結果,現存子孫がMMTVにもF-MuLVにも抵抗性となっているのではないかということである.実際,mA3bアリル産物N末端側の機能性アミノ酸配列多型に対しては,種の進化の過程で正の選択が加わっていた証拠がある38).
ところが,マウスAPOBEC3の分子進化に関する上記の仮説は,意外にも誤りであることが明らかとなった.ヒトの進化を人類発祥の地であるエチオピアからの移動の過程で追うことができるように,げっ歯類の進化も,それが最初に生まれたと考えられるインド亜大陸からの移動過程を追うことで推測できる.現存ネズミ科に属する野生マウス各種のAPOBEC3遺伝子エキソン5領域塩基配列およびその転写産物を互いに比較すると,驚くべきことに祖先型のAPOBEC3はΔ5型であり,現在のBALB/cやAマウスの祖先に当たる種が,カスピ海西岸から地中海沿岸に移動してアフリカ北部や西ヨーロッパに分布する過程で,エキソン5内部のSNPとイントロン4のスプライシングアクセプター部位多型を獲得して,全長型のAPOBEC3を発現するようになったと考えられた37).しかも,全長型を発現する種では,そのN末端側のアミノ酸配列は基本的に現在のmA3dアリルと同じであり,この部位のアミノ酸配列維持に強い負の選択圧が加わっている(図4).一方,カスピ海東岸を通ってシベリア西部に至り,現在の東ヨーロッパから北ヨーロッパに分布した祖先種ではΔ5型が維持され,そこに段階的にN末端側のアミノ酸配列置換が蓄積していった.そして,これとは独立に,ある時点でΔ5型を発現する祖先個体でイントロン2に異種指向性レトロウイルスのLTRが組込まれ,イントロンエンハンサーの獲得でΔ5が高発現となった個体の子孫が,進化上の優位性を獲得して現在のB6/B10系統の祖先となったと考えられる.Δ5型を発現する祖先種に,イントロンエンハンサー獲得とN末端アミノ酸置換に対する正の選択が加わって現在のmA3bアリルが生まれたことは想像にかたくないが,わざわざタンパク質低発現となるmA3dアリルが生まれ,しかもそのN末端側アミノ酸配列に対して強い維持圧力が加わっているのはなぜだろうか?
これについて興味深いのは,野生種マウスにおける染色体中の内在性異種指向性レトロウイルス分布43)と,Δ5型APOBEC3遺伝子の分布とがほぼ一致していることである.現在の野生型および実験室系マウスが染色体上に持つ内在性異種指向性ウイルスは,ラットを介して種間感染で他種生物に持ち込まれたものと考えられている44).現存の実験室系マウスは,その祖先が持っていた異種指向性ウイルスの受容体(XPR1)に多型を獲得しており,異種指向性ウイルスはその名のとおり実験室系統のマウスには感染できない43).しかし,機能性のXPR1を発現していたネズミ科の祖先が,カスピ海東岸からシベリア西部に移動する過程で,他種動物(ラット?)から繰り返して異種指向性レトロウイルスの攻撃を受けたことは間違いなく,その証拠としてこれら地域に分布する野生マウスとその子孫の実験室系統には,染色体上に多数の異種指向性ウイルスのプロウイルスが内在化している.
一方,地中海沿岸を西進したネズミ科の祖先に由来する現存野生マウスには,内在性異種指向性ウイルスが検出されない43).その一方で,これらのマウスの染色体中には多数の内在性多指向性ウイルスが存在する.多指向性ウイルスもXPR1を受容体とし,その被膜タンパク質は現存マウス系統のXPR1に結合できるが,内在性多指向性ウイルスのプロウイルスでは構造遺伝子の変異が蓄積しており,直接感染性粒子を作ることができない.内在性多指向性ウイルスが感染性を獲得するのは,AKVやMo-MuLVを新生仔に接種した場合のように,感染性の同種指向性ウイルスによるウイルス血症が持続し,組換え型ウイルスが形成される場合だけである.
興味深いことに,内在性多指向性ウイルスのプロウイルスには,APOBEC3によって生じたG-to-A変異が蓄積しているが,内在性異種指向性ウイルスのプロウイルスには,それがない45).このことは,Δ5型のマウスAPOBEC3がデアミナーゼ非依存性の複製抑制活性を示すこと35, 39)と符合する.すなわち,異種指向性ウイルスの複製抑制はデアミナーゼ活性に依存せず,APOBEC3タンパク質の発現量とN末端側のアミノ酸配列多型が異種指向性ウイルスに対する抵抗性発揮に重要であるのに対し,多指向性ウイルスの複製抑制にはデアミナーゼ活性が必須であり,そのためにはエキソン5の獲得とN末端側のアミノ酸配列をmA3d型に保つこととが有利であったと推測される.このような理論的推測の当否は,今後実験的に検証していかなければならない.
異種指向性レトロウイルスと多指向性レトロウイルスを選択圧と仮定してAPOBEC3遺伝子の機能的多型獲得の過程をたどることで,哺乳動物とレトロウイルスの相互作用による進化の過程が明らかにできるであろう.現存マウスの内在性多指向性ウイルスは複製能を失っているため,祖先型の感染性多指向性ウイルスを再構築する必要があるが,内在性多指向性プロウイルスの相互比較により,それは可能になると考える.
レトロウイルス抵抗性因子としてのマウスAPOBEC3を考えるとき,その多型がRfv3遺伝子同定の過程で見いだされたことは忘れてはならない.すなわち,mA3アリルの機能差が,抵抗性および感受性のRfv3遺伝子型を説明できるかを考える必要がある. Santiagoらは,APOBEC3多型はRfv3遺伝子現象そのものであると主張し46),APOBEC3がウイルス中和抗体の体細胞突然変異に関与すると考えている47).一方,我々はウイルス中和抗体産生に対するAPOBEC3遺伝子多型の効果は,ウイルス複製阻害を介する間接的な効果と考えており48),Santiagoらも以前はその説に同意していた49).また,我々はフレンドウイルスに対する中和抗体産生には抗体遺伝子可変部の体細胞突然変異は必要でないことを示している(むしろ,早期のIgM抗体産生が重要である)50).APOBEC3 mRNAの発現には細胞種間の差があり,Bリンパ球での発現がTリンパ球におけるそれよりも明らかに高いのは事実であるが48),APOBEC3が抗体産生における体細胞突然変異に関与するならば,Bリンパ球の核内にAPOBEC3が検出されなければならない.今後,生体内の各細胞種間におけるAPOBEC3分子の核内分布の比較と,ウイルス感染に伴うその変化の解析が慎重に行われる必要がある.また,FV感染後の中和抗体産生は明らかにT細胞依存性であり51),MHC遺伝子型(H2ハプロタイプ)によって抗体産生の速度は変わってくる52).Rfv3遺伝子とmA3アリルの異同を論じる場合に,H2ハプロタイプの異なるマウス間の比較はできないことを念頭に置く必要がある.
引用文献References
1) Prugnolle, F., Manica, A., Charpentier, M., Guégan, J.F., Guernier, V., & Balloux, F. (2005) Curr. Biol., 15, 1022–1027.
2) Duggal, N.K. & Emerman, M. (2012) Nat. Rev. Immunol., 12, 687–695.
3) Miyazawa, M., Tsuji-Kawahara, S., & Kanari, Y. (2008) Vaccine, 26, 2981–2996.
4) Perez-Caballero, D., Soll, S.J., & Bieniasz, P.D. (2008) PLoS Pathog., 4, e1000181.
5) Rose, K.M., Marin, M., Kozak, S.L., & Kabat, D. (2004) Trends Mol. Med., 10, 291–297.
6) Sheehy, A.M., Gaddis, N.C., Choi, J.D., & Malim, M.H. (2002) Nature, 418, 646–650.
7) Biasin, M., Piacentini, L., Lo Caputo, S., Kanari, Y., Magri, G., Trabattoni, D., Naddeo, V., Lopalco, L., Clivio, A., Cesana, E., Fasano, F., Bergamaschi, C., Mazzotta, F., Miyazawa, M., & Clerici, M. (2007) J. Infect. Dis., 195, 960–964.
8) Okeoma, C.M., Lovsin, N., Peterlin, B.M., & Ross, S.R. (2007) Nature, 445, 927–930.
9) Ross, S.R. (2010) Viruses, 2, 2000–2012.
10) Okeoma, C.M., Huegel, A.L., Lingappa, J., Feldman, M.D., & Ross, S.R. (2010) Cell Host Microbe., 16, 534–543.
11) Hoffman, P.M., Davidson, W.F., Ruscetti, S.K., Chused, T.M., & Morse, H.C. III. (1981) J. Virol., 39, 597–602.
12) Kabat, D. (1989) Curr. Top. Microbiol. Immunol., 148, 1–42.
13) Tsuji-Kawahara, S., Kawabata, H., Matsukuma, H., Kinoshita, S., Chikaishi, T., Sakamoto, M., Kawasaki, Y., & Miyazawa, M. (2013) J. Virol., 87, 13760–13774.
14) Best, S., Le Tissier, P., Towers, G., & Stoye, J.P. (1996) Nature, 382, 826–829.
15) Persons, D.A., Paulson, R.F., Loyd, M.R., Herley, M.T., Bodner, S.M., Bernstein, A., Correll, P.H., & Ney, P.A. (1999) Nat. Genet., 23, 159–165.
16) Ikeda, H. & Sugimura, H. (1989) J. Virol., 63, 5405–5412.
17) Chesebro, B., Miyazawa, M., & Britt, W.J. (1990) Annu. Rev. Immunol., 8, 477–499.
18) Kondo, T., Uenishi, H., Shimizu, T., Hirama, T., Iwashiro, M., Kuribayashi, K., Tamamura, H., Fujii, N., Fujisawa, R., Miyazawa, M., & Yamagishi, H. (1995) J. Virol., 69, 6735–6741.
19) Chen, W., Qin, H., Chesebro, B., & Cheever, M.A. (1996) J. Virol., 70, 7773–7782.
20) Takamura, S., Tsuji-Kawahara, S., Yagita, H., Akiba, H., Sakamoto, M., Chikaishi, T., Kato, M., & Miyazawa, M. (2010) J. Immunol., 184, 4696–4707.
21) Takamura, S., Kajiwara, E., Tsuji-Kawahara, S., Masumoto, T., Fujisawa, M., Kato, M., Chikaishi, T., Kawasaki, Y., Kinoshita, S., Itoi, M., Sakaguchi, N., & Miyazawa, M. (2014) PLoS Pathog., 10, e1003937.
22) Peterson, K.E., Stromnes, I., Messer, R., Hasenkrug, K., & Chesebro, B. (2002) J. Virol., 76, 7942–7948.
23) Miyazawa, M., Nishio, J., Wehrly. K., David, C. S., & Chesebro, B. (1992) J. Immunol., 148, 1964–1967.
24) Miyazawa, M., Nishio, J., & Chesebro, B. (1988) J. Exp. Med., 168, 1587–1605.
25) Iwashiro, M., Kondo, T., Shimizu, T., Yamagishi, H., Takahashi, K., Matsubayashi, Y., Masuda, T., Otaka, A., Fujii, N., Ishimoto, A., Miyazawa, M., Robertson, M.N., Chesebro, B., & Kuribayashi, K. (1993) J. Virol., 67, 4533–4542.
26) Sugahara, D., Tsuji-Kawahara, S., & Miyazawa, M. (2004) J. Virol., 78, 6322–6334.
27) Miyazawa, M., Fujisawa, R., Ishihara, C., Takei, Y.A., Shimizu, T., Uenishi, H., Yamagishi, H., & Kuribayashi, K. (1995) J. Immunol., 155, 748–758.
28) Kawabata, H., Niwa, A., Tsuji-Kawahara, S., Uenishi, H., Iwanami, N., Matsukuma, H., Abe, H., Tabata, N., Matsumura, H., & Miyazawa, M. (2006) Int. Immunol., 18, 183–198.
29) Chesebro, B. & Wehrly, K. (1979) Proc. Natl. Acad. Sci. USA, 76, 425–429.
30) Doig, D. & Chesebro, B. (1979) J. Exp. Med., 150, 10–19.
31) Hasenkrug, K.J., Valenzuela, A., Letts, V.A., Nishio, J., Chesebro, B., & Frankel, W.N. (1995) J. Virol., 69, 2617–2620.
32) Super, H.J., Hasenkrug, K.J., Simmons, S., Brooks, D.M., Konzek, R., Sarge, K.D., Morimoto, R.I., Jenkins, N.A., Gilbert, D.J., Copeland, N.G., Frankel, W., & Chesebro, B. (1999) J. Virol., 73, 7848–7852.
33) Kanari, Y., Clerici, M., Abe, H., Kawabata, H., Trabattoni, D., Lo Caputo, S., Mazzotta, F., Fujisawa, H., Niwa, A., Ishihara, C., Takei, Y.A., & Miyazawa, M. (2005) AIDS, 19, 1015–1024.
34) Santiago, M.L., Montano, M., Benitez, R., Messer, R.J., Yonemoto, W., Chesebro, B., Hasenkrug, K.J., & Greene, W.C. (2008) Science, 321, 1343–1346.
35) Takeda, E., Tsuji-Kawahara, S., Sakamoto, M., Langlois, M.A., Neuberger, M.S., Rada, C., & Miyazawa, M. (2008) J. Virol., 82, 10998–11008.
36) Okeoma, C.M., Petersen, J., & Ross, S.R. (2009) J. Virol., 83, 3029–3038.
37) Li, J., Hakata, Y., Takeda, E., Liu, Q., Iwatani, Y., Kozak, C.A., & Miyazawa, M. (2012) PLoS Pathog., 8, e1002478.
38) Sanville, B., Dolan, M.A., Wollenberg, K., Yan, Y., Martin, C., Yeung, M.L., Strebel, K., Buckler-White, A., & Kozak, C.A. (2010) PLoS Pathog., 6, e1000974.
39) Okeoma, C.M., Low, A., Bailis, W., Fan, H.Y., Peterlin, B.M., & Ross, S.R. (2009) J. Virol., 83, 3486–3495.
40) Abudu, A., Takaori-Kondo, A., Izumi, T., Shirakawa, K., Kobayashi, M., Sasada, A., Fukunaga, K., & Uchiyama, T. (2006) Curr. Biol., 16, 1565–1570.
41) MacMillan, A.L., Kohli, R.M., & Ross, S.R. (2013) J. Virol., 87, 4808–4817.
42) Langlois, M.A., Kemmerich, K., Rada, C., & Neuberger, M.S. (2009) J. Virol., 83, 11550–11559.
43) Kozak, C.A. (2010) Retrovirology, 7, 101.
44) Hayward, A., Grabherr, M., & Jern, P. (2013) Proc. Natl. Acad. Sci. USA, 110, 20146–20151.
45) Jern, P., Stoye, J.P., & Coffin, J.M. (2007) PLoS Genet., 3, 2014–2022.
46) Santiago, M.L., Smith, D.S., Barrett, B.S., Montano, M., Benitez, R.L., Pelanda, R., Hasenkrug, K.J., & Greene, W.C. (2011) J. Virol., 85, 189–199.
47) Halemanoa, K., Guoa, K., Heilmana, K.J., Barretta, B.S., Smitha, D.S., Hasenkrugc, K.J., & Santiago, M.L. (2014) Proc. Natl. Acad. Sci. USA, 111, 7759–7764.
48) Tsuji-Kawahara, S., Chikaishi, T., Takeda, E., Kato, M., Kinoshita, S., Kajiwara, E., Takamura, S., & Miyazawa, M. (2010) J. Virol., 84, 6082–6095.
49) Santiago, M.L., Benitez, R.L., Montano, M., Hasenkrug, K.J., & Greene, W.C. (2010) J. Immunol., 185, 1114–1123.
50) Kato, M., Tsuji-Kawahara, S., Kawasaki, Y., Kinoshita, S., Chikaishi, T., Takamura, S., Fujisawa, M., Kawada, A., & Miyazawa, M. (2015) J. Virol., 89, 1468–1473.
51) Super, H.J., Brooks, D., Hasenkrug, K., & Chesebro, B. (1998) J. Virol., 72, 9400–9403.
52) Miyazawa, M., Nishio, J., Wehrly, K., & Chesebro, B. (1992) J. Immunol., 148, 644–647.
著者紹介Author Profile
 宮澤 正顯(みやざわ まさあき)
宮澤 正顯(みやざわ まさあき)近畿大学医学部教授(免疫学教室主任),近畿大学医学部共同研究施設長.医学博士(東北大学).
略歴1982年東北大学医学部卒業,医師免許証取得.84年東北大学助手(医学部病理学講座).86年アメリカ合衆国NIH, NIAID Visiting Associate.87年医学博士.90年東北大学助手復職.95年三重大学医学部助教授(生体防御医学講座).96年近畿大学医学部教授.現在に至る.
研究テーマと抱負レトロウイルス感染に対する宿主免疫応答の遺伝子支配機構を解明し,感染防御へと繋げること
ウェブサイトhttp://www.med.kindai.ac.jp/immuno/
趣味クラシック音楽,庭の木々と芝生の手入れ.