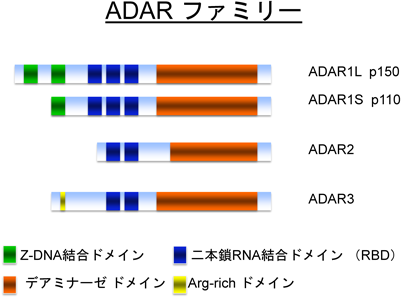
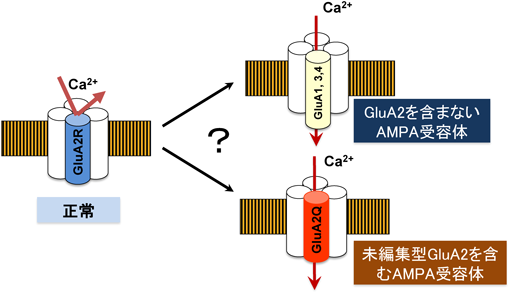
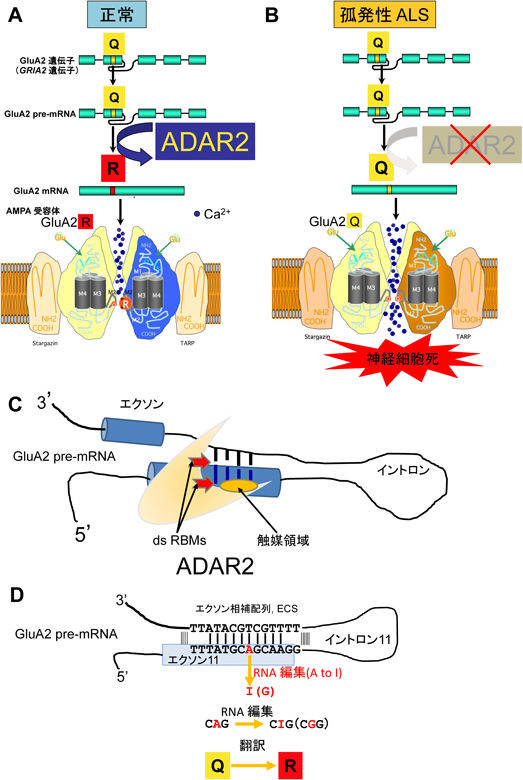
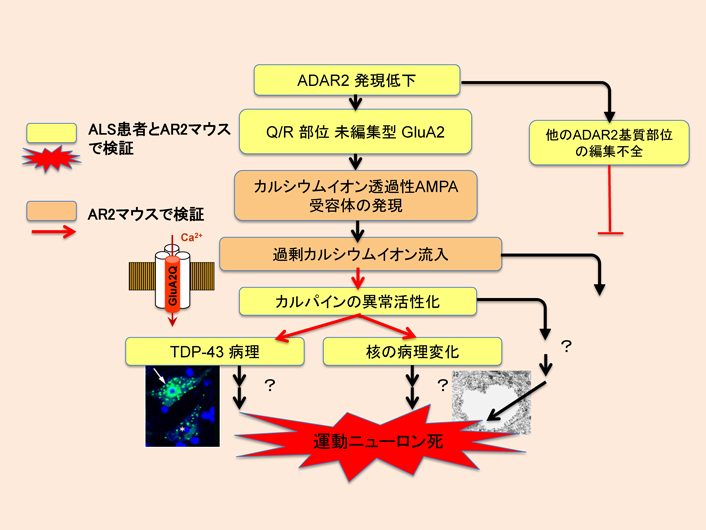
11) Cirulli, E.T., Lasseigne, B.N., Petrovski, S., Sapp, P.C., Dion, P.A., Leblond, C.S., Couthouis, J., Lu, Y.F., Wang, Q., Krueger, B.J., Ren, Z., Keebler, J., Han, Y., Levy, S.E., Boone, B.E., Wimbish, J.R., Waite, L.L., Jones, A.L., Carulli, J.P., Day-Williams, A.G., Staropoli, J.F., Xin, W.W., Chesi, A., Raphael, A.R., McKenna-Yasek, D., Cady, J., Vianney de Jong, J.M., Kenna, K.P., Smith, B.N., Topp, S., Miller, J., Gkazi, A., Al-Chalabi, A., van den Berg, L.H., Veldink, J., Silani, V., Ticozzi, N., Shaw, C.E., Baloh, R.H., Appel, S., Simpson, E., Lagier-Tourenne, C., Pulst, S.M., Gibson, S., Trojanowski, J.Q., Elman, L., McCluskey, L., Grossman, M., Shneider, N.A., Chung, W.K., Ravits, J.M., Glass, J.D., Sims, K.B., Van Deerlin, V.M., Maniatis, T., Hayes, S.D., Ordureau, A., Swarup, S., Landers, J., Baas, F., Allen, A.S., Bedlack, R.S., Harper, J.W., Gitler, A.D., Rouleau, G.A., Brown, R., Harms, M.B., Cooper, G.M., Harris, T., Myers, R.M., & Goldstein, D.B.; FALS Sequencing Consortium. (2015) Science, 347, 1436–1441.
13) Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., Clark, C.M., McCluskey, L.F., Miller, B.L., Masliah, E., Mackenzie, I.R., Feldman, H., Feiden, W., Kretzschmar, H.A., Trojanowski, J.Q., & Lee, V.M. (2006) Science, 314, 130–133.
19) Renton, A.E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J.R., Schymick, J.C., Laaksovirta, H., van Swieten, J.C., Myllykangas, L., Kalimo, H., Paetau, A., Abramzon, Y., Remes, A.M., Kaganovich, A., Scholz, S.W., Duckworth, J., Ding, J., Harmer, D.W., Hernandez, D.G., Johnson, J.O., Mok, K., Ryten, M., Trabzuni, D., Guerreiro, R.J., Orrell, R.W., Neal, J., Murray, A., Pearson, J., Jansen, I.E., Sondervan, D., Seelaar, H., Blake, D., Young, K., Halliwell, N., Callister, J.B., Toulson, G., Richardson, A., Gerhard, A., Snowden, J., Mann, D., Neary, D., Nalls, M.A., Peuralinna, T., Jansson, L., Isoviita, V.M., Kaivorinne, A.L., Holtta-Vuori, M., Ikonen, E., Sulkava, R., Benatar, M., Wuu, J., Chio, A., Restagno, G., Borghero, G., Sabatelli, M., Heckerman, D., Rogaeva, E., Zinman, L., Rothstein, J.D., Sendtner, M., Drepper, C., Eichler, E.E., Alkan, C., Abdullaev, Z., Pack, S.D., Dutra, A., Pak, E., Hardy, J., Singleton, A., Williams, N.M., Heutink, P., Pickering-Brown, S., Morris, H.R., Tienari, P.J., & Traynor, B.J.; ITALSGEN Consortium (2011) Neuron, 72, 257–268.
20) DeJesus-Hernandez, M., Mackenzie, I.R., Boeve, B.F., Boxer, A.L., Baker, M., Rutherford, N.J., Nicholson, A.M., Finch, N.A., Flynn, H., Adamson, J., Kouri, N., Wojtas, A., Sengdy, P., Hsiung, G.Y., Karydas, A., Seeley, W.W., Josephs, K.A., Coppola, G., Geschwind, D.H., Wszolek, Z.K., Feldman, H., Knopman, D.S., Petersen, R.C., Miller, B.L., Dickson, D.W., Boylan, K.B., Graff-Radford, N.R., & Rademakers, R. (2011) Neuron, 72, 245–256.
22) Donnelly, C.J., Zhang, P.W., Pham, J.T., Heusler, A.R., Mistry, N.A., Vidensky, S., Daley, E.L., Poth, E.M., Hoover, B., Fines, D.M., Maragakis, N., Tienari, P.J., Petrucelli, L., Traynor, B.J., Wang, J., Rigo, F., Bennett, C.F., Blackshaw, S., Sattler, R., & Rothstein, J.D. (2013) Neuron, 80, 415–428.
31) Kwiatkowski, T.J. Jr., Bosco, D.A., Leclerc, A.L., Tamrazian, E., Vanderburg, C.R., Russ, C., Davis, A., Gilchrist, J., Kasarskis, E.J., Munsat, T., Valdmanis, P., Rouleau, G.A., Hosler, B.A., Cortelli, P., de Jong, P.J., Yoshinaga, Y., Haines, J.L., Pericak-Vance, M.A., Yan, J., Ticozzi, N., Siddique, T., McKenna-Yasek, D., Sapp, P.C., Horvitz, H.R., Landers, J.E., & Brown, R.H. Jr. (2009) Science, 323, 1205–1208.
32) Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K.J., Nishimura, A.L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P., Ganesalingam, J., Williams, K.L., Tripathi, V., Al-Saraj, S., Al-Chalabi, A., Leigh, P.N., Blair, I.P., Nicholson, G., de Belleroche, J., Gallo, J.M., Miller, C.C., & Shaw, C.E. (2009) Science, 323, 1208–1211.
34) Mackenzie, I.R., Bigio, E.H., Ince, P.G., Geser, F., Neumann, M., Cairns, N.J., Kwong, L.K., Forman, M.S., Ravits, J., Stewart, H., Eisen, A., McClusky, L., Kretzschmar, H.A., Monoranu, C.M., Highley, J.R., Kirby, J., Siddique, T., Shaw, P.J., Lee, V.M., & Trojanowski, J.Q. (2007) Ann. Neurol., 61, 427–434.
40) Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., Beach, T.G., Buratti, E., Baralle, F., Morita, M., Nakano, I., Oda, T., Tsuchiya, K., & Akiyama, H. (2008) Ann. Neurol., 64, 60–70.
44) Zhang, K., Donnelly, C.J., Haeusler, A.R., Grima, J.C., Machamer, J.B., Steinwald, P., Daley, E.L., Miller, S.J., Cunningham, K.M., Vidensky, S., Gupta, S., Thomas, M.A., Hong, I., Chiu, S.L., Huganir, R.L., Ostrow, L.W., Matunis, M.J., Wang, J., Sattler, R., Lloyd, T.E., & Rothstein, J.D. (2015) Nature, 525, 56–61.
45) Freibaum, B.D., Lu, Y., Lopez-Gonzalez, R., Kim, N.C., Almeida, S., Lee, K.H., Badders, N., Valentine, M., Miller, B.L., Wong, P.C., Petrucelli, L., Kim, H.J., Gao, F.B., & Taylor, J.P. (2015) Nature, 525, 129–133.
46) Kaneb, H.M., Folkmann, A.W., Belzil, V.V., Jao, L.E., Leblond, C.S., Girard, S.L., Daoud, H., Noreau, A., Rochefort, D., Hince, P., Szuto, A., Levert, A., Vidal, S., Andre-Guimont, C., Camu, W., Bouchard, J.P., Dupre, N., Rouleau, G.A., Wente, S.R., & Dion, P.A. (2015) Hum. Mol. Genet., 24, 1363–1373.
53) Corcia, P. & Gordon, P.H. (2012) Ther. Clin. Risk Manag., 8, 359–366.