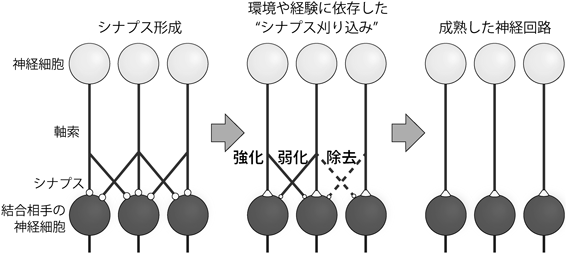
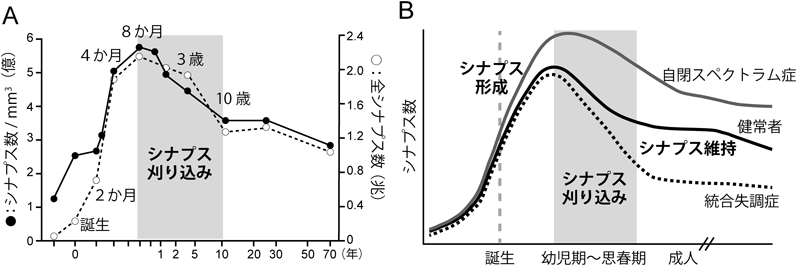
生まれたばかりの動物の神経系では,盛んにシナプス形成が起こり,その密度は成熟動物の神経系よりもずっと高くなるが,一般に,出生直後に形成されたシナプスは機能的に未熟であり,動物個体としても脳機能は未熟な状態にある.成長につれて,必要なシナプスは強められて残存し,不必要なシナプスは弱められ最終的に除去される.この過程は「シナプス刈り込み」と呼ばれており,哺乳類や爬虫類などさまざまな動物種でみられ,神経系のさまざまな部位のシナプスで起こることから,生後発達期の神経系で普遍的に起こる重要な現象であり,成熟した機能的神経回路を作るための基本的過程であると考えられている1–4)(図1).たとえばヒトの大脳皮質視覚野の場合,生後8か月齢までに盛んにシナプス形成が起こるが,その後,発達に伴ってシナプス密度は徐々に減少していき,10歳ごろまでにほぼ半減する.それ以降は,老化による細胞死が顕著になるまでの数十年の間,シナプス密度はほぼ一定に維持される5)(図2A).シナプス刈り込みは,体性感覚系,聴覚系,視覚系,運動系など,神経系のさまざまな部位でみられるが,なかでも,小脳の登上線維–プルキンエ細胞シナプスの刈り込みはその過程や分子メカニズムが最もよく研究されており,末梢神経系の神経筋接合部とならんでシナプス刈り込みの代表的なモデル実験系として,広く認知されている.最近では,シナプス刈り込みの異常が自閉スペクトラム症や統合失調症といった精神神経疾患との関連においても注目されている6)(図2B).本稿では,小脳の登上線維–プルキンエ細胞シナプスの刈り込みに関してその過程と分子メカニズムを概説するとともに,我々の最近の成果を紹介したい.
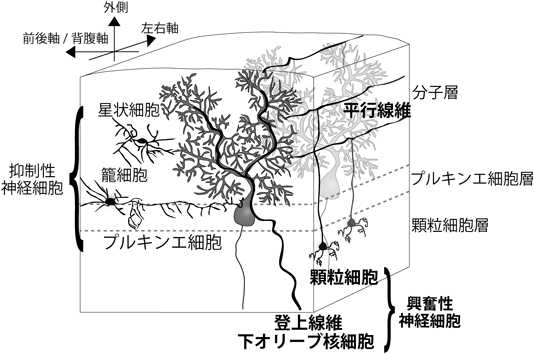
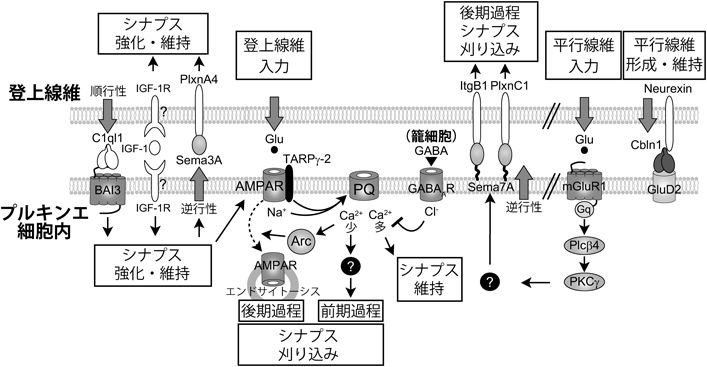
小脳のプルキンエ細胞は,グルタミン酸を伝達物質とする2種類の興奮性入力を樹状突起の異なる領域で受け取る(図3).一つの入力は小脳皮質の顆粒細胞の軸索である平行線維からの入力で,プルキンエ細胞の遠位樹状突起のスパインにシナプスを形成している.個々のプルキンエ細胞にはマウスでは10万本の平行線維がシナプス結合しているが,それぞれの入力の強さは弱い.顆粒細胞は橋,延髄,脊髄にあるさまざまな神経核から苔状線維を介して興奮性入力を受けている.もう一つの入力は延髄の下オリーブ核神経細胞の軸索である登上線維からの入力である.成熟した動物では,一つのプルキンエ細胞には1本の登上線維のみが樹状突起の近位部にシナプスを形成している.1本の登上線維は一つのプルキンエ細胞に数百のシナプス結合を持ち,さらにシナプス後部のAMPA型グルタミン酸受容体の密度も平行線維と比べて高いため,登上線維の入力はプルキンエ細胞に非常に強い脱分極を引き起こす.プルキンエ細胞は興奮性入力だけでなく,GABAを伝達物質とする抑制性入力も受けている.分子層のうちでプルキンエ細胞層に近い深層に存在する籠細胞はプルキンエ細胞の細胞体に,分子層中間層に存在する星状細胞はプルキンエ細胞の樹状突起に,それぞれシナプスを作っている.
登上線維–プルキンエ細胞シナプスの発達過程について,最初は麻酔下のラットにおいてin vivoで電気生理学的解析が行われた.プルキンエ細胞から記録をして登上線維への電気刺激の強度を徐々に強くしていくと,成熟動物では,全か無かの法則に従って単一のシナプス応答がみられるのに対し,幼若動物では,段階的な複数のシナプス応答が誘発された7).この発見は,成熟動物では個々のプルキンエ細胞が1本の強い登上線維からのみ入力を受けており,幼若動物では複数の登上線維から入力を受けていることを意味している.その後の研究により,幼若期から成熟するにつれ個々のプルキンエ細胞に結合する登上線維の数が減少し,最終的にはほとんどのプルキンエ細胞は1本の登上線維からのみ入力を受けるようになることがわかった.
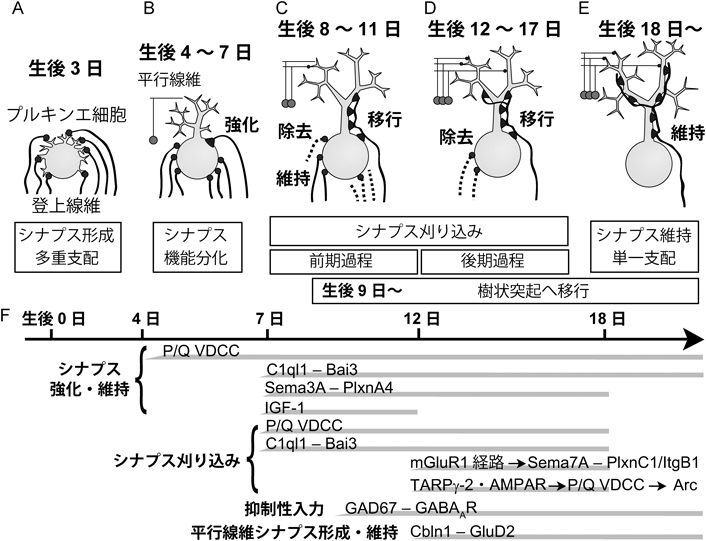
その後,形態学的解析が行われ,また急性スライス標本からのホールセル(全細胞)記録による詳細な電気生理学的解析が行われて,登上線維–プルキンエ細胞シナプスの発達過程が明らかにされた(図4).出生直後,プルキンエ細胞は複数の小さな樹状突起を持ち,登上線維は最初その小さな樹状突起にシナプスを形成する8).少し発達が進み,プルキンエ細胞の樹状突起が退縮するにつれ,登上線維のシナプスはプルキンエ細胞の細胞体に認められるようになる.生後3日のマウスでは,プルキンエ細胞は複数の登上線維からほぼ同じ強さのシナプス入力を受け取っている9)(図4A).生後7日までに,プルキンエ細胞から1本の樹状突起が伸び出すとともに,細胞体における登上線維シナプスの密度が増加し,個々のプルキンエ細胞は平均約7本の登上線維からシナプス入力を受けるようになる.生後4日から7日の間に,1本の登上線維からのシナプス入力が,他の登上線維からの入力に比べ選択的に強くなるが,この時期には登上線維シナプスは細胞体にある.この過程は“機能分化”と呼ばれている9)(図4B).生後9日ごろから,選択的に強化された1本の登上線維だけが,プルキンエ細胞の細胞体から樹状突起へとシナプスを形成し始め,生後20日には分子層の長さの約75%の位置まで,その名のとおり,登上する10)(図4C~E).
登上線維–プルキンエ細胞シナプスの刈り込みは,細胞体に残存している登上線維シナプスに起こる(図4C~E).生後7日までの単一登上線維の選択的強化の間,プルキンエ細胞にシナプス入力をする登上線維の数にほとんど変化がないことから,この間は登上線維の正味の除去は起こらず,1本の強い登上線維の他に平均6本の弱い登上線維のシナプスが細胞体に残されていると考えられる.強化された登上線維の樹状突起への移行と平行して,生後17日ごろまでに,細胞体に残されたシナプスが徐々に除去されていく.この除去過程には,“前期過程”と“後期過程”の少なくとも2段階あり,生後8日から11日あたりで起こる前期過程でおよそ4本(図4C),生後12日ごろからの後期過程で2本ほどが除去される11, 12)(図4D).すなわち,平行線維–プルキンエ細胞シナプス形成とは無関係に起こる前期過程と,正常な平行線維–プルキンエ細胞シナプス形成が前提条件となる後期過程に分かれる(図4F).このことは,デルタ2グルタミン酸受容体GluRδ2(GluD2)が欠損したマウスの解析により明らかにされた11).GluD2は,Cbln1と結合する膜タンパク質であり(後述),プルキンエ細胞樹状突起の平行線維シナプス後部に局在している.GluD2欠損マウスでは,平行線維のシナプス形成に異常がみられ,正常に比べてシナプス数は約半分に減少している13, 14).登上線維シナプスの発達に関しては,生後11日までは目立った変化はみられなかったが,生後12日からプルキンエ細胞にシナプス入力する登上線維の数が野生型マウスと比べて有意に多かった11).すなわちシナプスの刈り込みには生後8日から11日ごろまでの前期過程と12日から17日ごろまでの後期過程があり,前期過程は平行線維シナプス形成に依存せず,後期過程は平行線維シナプスの正常な形成が必要であると結論された(図4).
3. シナプス刈り込みの機能分化と前期過程のメカニズム
P/Q型電位依存性Ca2+チャネル(P/Q-type voltage-dependent calcium channel:P/Q VDCC)Cacna1a/Cav2.1は,プルキンエ細胞の樹状突起と細胞体にCa2+電流を発生させる主要なVDCCである.このP/Q VDCCを全身でノックアウトしたマウスでは,シナプス刈り込みが障害されている15).我々は,P/Q VDCCのプルキンエ細胞特異的ノックアウトマウスを作製して,登上線維–プルキンエ細胞シナプスの刈り込み過程を精査した16).このノックアウトマウスでは,生後4日から7日に起こる機能分化が著しく障害されていた.また,生後6日までは個々のプルキンエ細胞を支配する登上線維の数は正常であったが,生後7日から野生型マウスに比べて登上線維の数が有意に多くなり,生後20日過ぎまでこの差は顕著であったことから,登上線維シナプス除去の前期過程が障害されていることが判明した.さらに,このノックアウトマウスでは2本以上の登上線維が樹状突起へ移行していた.したがって,プルキンエ細胞のP/Q VDCCは機能分化,シナプス除去の前期過程,単一登上線維の樹状突起への移行に必須であることが明らかとなった(図4F).
インスリン様成長因子I(insulin-like growth factor I:IGF-1)と脳由来神経栄養因子(BDNF)の受容体であるTrkBは,シナプス除去の前期過程に関与している可能性が示されている.Elvax樹脂を用いてIGF-1タンパク質を生後8日から小脳に投与したとき,生後13日で,強い登上線維シナプスに関しては変化がなかったが,相対的に弱い登上線維シナプスの入力が対照群に比べて大きくなり,しかも多くの登上線維が残っていることがわかった17).また,抗IGF抗体を生後8日から小脳に投与したときは,生後13日で,逆に,強い登上線維シナプスの入力が小さくなり,登上線維が少なくなる.これらの結果から,IGF-1は生後12日までに起こる登上線維入力の強化とシナプス除去の前期過程に関与することが示唆されている(図4F).IGF-1投与実験の結果と似たような表現型がTrkBの小脳ノックアウトマウスでみられていて,通常のマウスと比べ登上線維間での入力強度に差がなく,生後12日の段階で個々のプルキンエ細胞を支配する登上線維の数が多いと報告されている18, 19).
登上線維間で入力強度に差を生じさせるメカニズムとして,生後1週目ごろにプルキンエ細胞と登上線維の同期活動により長期増強や長期抑圧が引き起こされることが示唆されている20, 21).同じ同期活動でも入力強度が強い登上線維では長期増強が起こり,入力強度が弱い登上線維では長期抑圧が起こる.これにより強い登上線維はより強くなり,弱い登上線維はより弱くなり最終的に除去されるメカニズムが考えられる.また,生体内のプルキンエ細胞で自発神経活動を観察した我々の研究から,プルキンエ細胞の活動とより近いタイミングで活動した登上線維が選択的に強化される可能性が示された22).
前述したように正常な平行線維のシナプス形成が,生後12日以降の登上線維のシナプス刈り込みの後期過程の前提条件である.平行線維のシナプス形成と維持に重要なGluD2は,そのN末端ドメインにより平行線維からの分泌性タンパク質Cbln1を介して平行線維のNeurexinと相互作用し,この3分子が複合体を形成することが明らかとなった23, 24)(図4F,図5).GluD2およびCbln1ノックアウトマウスでは,シナプス除去が障害されており,登上線維がプルキンエ細胞樹状突起の遠位部にまで異所性のシナプスを形成する25–28).
代謝型グルタミン酸受容体mGluR1はプルキンエ細胞で強く発現しており,mGluR1からのシグナリングは登上線維–プルキンエ細胞シナプスの刈り込みに必須である29)(図4F).mGluR1とその下流因子であるGαq,ホスホリパーゼCβ4 (PLCβ4),タンパク質キナーゼCγ (PKCγ)のそれぞれのノックアウトマウスは,生後14日ごろまでは登上線維の数に異常はみられず,それ以降に過剰登上線維が残存していることから,mGluR1シグナリングはシナプス刈り込みの後期過程に関与することが示された30–32)(図5).これらのノックアウトマウスでは,小脳の構造,プルキンエ細胞の形態,平行線維シナプスは正常であることから,Cbln1–GluD2の作用とは異なり,mGluR1シグナリングが直接,過剰な弱い登上線維のシナプスの除去を引き起こすと考えられる.また,PLCβ4は小脳の領域特異的な発現パターンを示すが,PLCβ4のノックアウトマウスでは,PLCβ4が発現する領域のプルキンエ細胞でシナプス刈り込みの障害が観察された32).さらに,PKC阻害ペプチドをプルキンエ細胞でのみ発現させたマウスでもPKCγのノックアウトマウスと同様シナプス刈り込みが障害されていた33).これらの結果からmGluR1とその下流で働くPKCγなどの分子はプルキンエ細胞で機能することでシナプス刈り込みの後期過程を制御することが明らかとなった.
NMDA受容体は,発達期のプルキンエ細胞ではほとんど発現しておらず発達期の小脳では苔状線維–顆粒細胞シナプスで多く発現している.我々は,生後発達期のマウス小脳においてNMDA受容体を阻害する実験を行った.その結果,シナプス刈り込みの後期過程に障害がみられた34).登上線維–プルキンエ細胞シナプスや平行線維–プルキンエ細胞シナプスではNMDA受容体を介したシナプス伝達は検出されなかったことから,苔状線維–顆粒細胞シナプスのNMDA受容体がシナプス刈り込みの後期過程を制御する可能性が考えられている.mGluR1シグナリングが後期過程に必須であることを考え合わせると,苔状線維–顆粒細胞–平行線維を介する活動がプルキンエ細胞の樹状突起のmGluR1を活性化しGαq–PLCβ4-PKCγという一連の分子カスケードがプルキンエ細胞内で働くことが,後期過程における過剰登上線維シナプスの除去につながると結論された.
プルキンエ細胞は籠細胞と星状細胞から抑制性入力を受けている.我々は,GABAの合成が低下するよう遺伝子改変されたGAD67ヘテロ欠損マウスを用い,プルキンエ細胞への抑制性入力とシナプス刈り込みを解析した35).GAD67ヘテロ欠損マウスでは,生後7日から抑制性入力が減弱しており,さらにシナプス刈り込みの障害がみられ,生後10日以降で明白であった(図4F).その抑制性入力を詳細に解析した結果,籠細胞に由来する入力が減弱していることが明らかとなった.さらに,プルキンエ細胞で発生するCa2+濃度変化を解析した結果,強い登上線維を刺激したときに近位樹状突起で発生するCa2+濃度上昇はGAD67ヘテロ欠損マウスと野生型マウスとで差はなかったが,弱い登上線維を刺激したときに細胞体で発生するCa2+濃度上昇はGAD67ヘテロ欠損マウスの方が有意に大きかった.これらの結果は,弱い登上線維の除去にP/Q VDCCを介したCa2+流入が必要であることと一見矛盾するが,Ca2+上昇によって活性化される過程に,登上線維シナプスの除去を促進する過程と,逆に除去に対抗してシナプスを維持する過程があることを想定するとよく理解できる.GAD67ヘテロ欠損マウスではプルキンエ細胞の細胞体にある抑制性入力が減弱したため,弱い登上線維においても細胞体で過剰なCa2+が流入してしまい,シナプスを維持する過程が強く活性化されてしまうために,本来刈り込まれるべきシナプスが維持されてしまうと考えられる(図5).
6. 活動依存的に発現する初期応答遺伝子Arc/Arg3.1
プルキンエ細胞の神経活動がシナプス刈り込みに重要であることはP/Q VDCCのノックアウトマウスから示唆されている.さらに,プルキンエ細胞でCl−チャネルを過剰発現してその活動を抑制したマウスにおいて,シナプス刈り込みが障害されていた36).これらの研究はプルキンエ細胞の活動とCa2+流入が登上線維シナプスの刈り込みに必須であることを示しているが,Ca2+の下流で過剰登上線維を除去する働きをする分子に関してはまったく不明であった.
初期応答遺伝子Arc/Arg3.1 (Arc)は神経活動やCa2+流入に依存して発現が増加する遺伝子であり,これまでに長期増強や長期抑圧に関わることが示されていた37).しかし,Arcが発達期の神経回路形成に果たす役割は知られていなかった.我々はまずArcの発現が発達期に変化するかを調べた38).その結果,登上線維シナプスが刈り込まれている生後9日から16日にかけて小脳におけるArcのmRNAの量が顕著に増加することがわかった.次にプルキンエ細胞においてArcの発現が神経活動とP/Q VDCCによるCa2+流入に依存するかを調べた.小脳あるいはプルキンエ細胞を脱分極させた結果,プルキンエ細胞でArcの発現が有意に増加した.この増加は刺激時にP/Q VDCCを阻害すると起こらないことから,Arcの発現はプルキンエ細胞の神経活動ならびにP/Q VDCCを介したCa2+流入に依存することが明らかとなった.Arcがシナプス刈り込みに関与するかを調べるために,プルキンエ細胞でのみ働くL7プロモータによってArcに対するmiRNAを駆動するレンチウイルスベクターを用いた.生後1日から3日のマウスの小脳にArcのノックダウン配列を含むレンチウイルスを投与し,シナプス刈り込みの前期過程と後期過程を調べた.その結果,生後11日まではシナプス刈り込みに異常はみられなかったが,生後12日以降のシナプス刈り込みに障害がみられた(図4F).さらに,P/Q VDCCとArcをプルキンエ細胞で同時にノックダウンしたときにシナプス刈り込み異常は加算的にならず,Arc単独ノックダウン,P/Q VDCC単独ノックダウン,P/Q VDCCとArcのダブルノックダウンで同程度にシナプス刈り込みが障害されていた.これにより,ArcはP/Q VDCCの下流で働くことが示された.次に,P/Q VDCCのノックダウンとともにArcを過剰発現させて,シナプス刈り込み障害が回復するかどうかを調べたが,その障害は回復されなかった.このことはP/Q VDCCの下流でArcとともに働く別の分子があることを示唆している.以上より,P/Q VDCCはシナプス刈り込みの後期過程にも関与すること,プルキンエ細胞の神経活動ならびにP/Q VDCCの下流においてArcが発現し過剰な登上線維シナプスを除去すること,この除去にはArcとともに働く他のCa2+依存性の分子も必要なことが示唆された(図5).
前述したように,シナプス刈り込みにはシナプス後部の神経細胞の活動と分子カスケードが必要なことが証明されてきたが,そのような分子がどのようにシナプス前部の登上線維に働きかけ,シナプスを強化および維持あるいは除去するのかは不明のままであった.シナプス後部の分子が,逆行性シグナルとしてシナプス前部に直接働く可能性が考えられていたが,その実体は不明であった.我々は,この逆行性シグナル分子の候補を探索するため,まず,シナプス刈り込みが起こっている生後4日から15日の時期にプルキンエ細胞で発現する分子をマイクロアレイによりプロファイリングした.これらの分子の中で,逆行性シグナル分子として働きうる分泌性分子と膜分子に注目した.これらの分子がシナプス刈り込みに果たす役割を網羅的に調べるために,登上線維の起始核である下オリーブ核を含む延髄と小脳の共培養系の開発を行った39).この共培養系では登上線維シナプスがプルキンエ細胞に形成され,その後登上線維の選択的強化と過剰な登上線維の除去が起こる.さらに受容体の阻害剤やレンチウイルスによるRNA干渉ノックダウン法を用いGluD2, mGluR1, P/Q VDCC, NMDA受容体の関与を調べた結果,共培養系においてもこれらの分子がシナプス刈り込みに関わることが明らかとなった.
この共培養系を用い,候補分子をスクリーニングした.60種類以上の遺伝子を解析した結果,セマフォリン分子Semaphorin3A(Sema3A)とSemaphorin7A(Sema7A)がシナプス刈り込みに関与することが示された40).興味深いことに,Sema3Aをノックダウンすると登上線維のシナプス入力が減弱するとともに,シナプス刈り込みが促進される.一方,Sema7Aをノックダウンするとシナプス刈り込みが障害された.次に,生体内でいつ,どのようにSema3AとSema7Aがシナプス刈り込みを制御しているかを,レンチウイルスを介したRNA干渉ノックダウン法で調べた.その結果,Sema3Aのノックダウンにより,生後8日からシナプス刈り込みが促進され,さらに登上線維シナプスの興奮性シナプス後電流の振幅が減弱した.一方,Sema7Aのノックダウンでは,生後15日からシナプス刈り込みが障害された.これらの結果から,Sema3Aは生後8日から登上線維シナプスを強化・維持し,Sema7Aは生後15日から登上線維のシナプス除去を促進していることが明らかとなった(図4F).
前述したように,mGluR1, P/Q VDCC, GluD2が登上線維シナプス除去を促進する役割を果たすことが示されている.これらの分子とSema7Aとの関係を調べるために,mGluR1, P/Q VDCC, GluD2の中のいずれか一つとSema7Aのダブルノックダウンを行った.P/Q VDCCあるいはGluD2の一方とSema7Aをダブルノックダウンした場合には,単独ノックダウンと比べて加算的なシナプス刈り込み異常が観察された.一方,mGluR1とSema7Aをダブルノックダウンすると,単独ノックダウンと同じ効果が観察された.これらの結果から,Sema7AはmGluR1と同じシグナル経路にある可能性が示唆された.mGluR1ノックアウトマウスにおいて,Sema7Aタンパク質の発現が有意に減少していることが判明し,プルキンエ細胞においてmGluR1ノックダウンと同時にSema7Aの過剰発現をすると,mGluR1ノックダウンによる刈り込み障害が回復した.これらの結果より,Sema7A分子の発現がmGluR1により調節され,Sema7Aが登上線維シナプスを除去することが明らかになった(図4F,図5).
次に,Sema3AとSema7Aが直接プルキンエ細胞から登上線維に働きかける逆行性シグナルであるかを調べるために,登上線維においてそれらの受容体であるPlexin A4(PlxnA4),Integrin B1 (ItgB1),Plexin C1 (PlxnC1)の発現を,同様のノックダウン法で調べた.その結果,登上線維で,PlxnA4をノックダウンしたときはSema3Aと同じくシナプス刈り込みが促進され,ItgB1やPlxnC1をノックダウンしたときはSema7Aと同じくシナプス刈り込みが障害された.これらのリガンドと受容体の組み合わせをそれぞれプルキンエ細胞と登上線維で同時にノックダウンしても単独ノックダウンと同程度の異常しかみられないことから,これらは逆行性に直接シグナルを伝えていることが示唆された.これらにより,登上線維シナプスの強化・維持と除去が逆行性シグナルにより制御され,その分子実体として二つのセマフォリン分子Sema3AとSema7Aによるシグナルがあることが明らかとなった(図5).
8. AMPA受容体の補助因子TARPγ-2/Stargazinの役割
登上線維–プルキンエ細胞のシナプス伝達がシナプス刈り込みに果たす役割を明らかにするために,シナプス後部のAMPA型グルタミン酸受容体の量を制御しているTARPγ-2/Stargazinがプルキンエ細胞で欠損したマウスを解析した41).このマウスでは登上線維のシナプス刈り込みが進行する生後発達期において,登上線維–プルキンエ細胞のシナプス伝達が半分程度に減弱していた.一方,強い登上線維によるシナプス入力と弱い登上線維によるシナプス入力の相対的な強さの差は正常なままであった.次に,登上線維–プルキンエ細胞シナプスの刈り込みを解析した結果,生後7から11日では過剰登上線維のシナプス刈り込みが正常に進行したのに対して,生後12日からのシナプス刈り込みが障害されていた(図4F).また,このマウスでは強い登上線維のプルキンエ細胞樹状突起への移行が有意に低下していた.これらの結果から,生後7日から11日に起こるシナプス刈り込みの前期過程と登上線維のプルキンエ細胞樹状突起への伸展には,登上線維–プルキンエ細胞シナプスの伝達の絶対的な強さは影響しないのに対し,生後12日から起こる後期過程においては,個々のシナプス結合の絶対的な強さが重要であることが明らかとなった.さらに,シナプス刈り込みの後期過程に異常がみられたメカニズムとしてArcに注目した.このTARPγ-2ノックアウトマウスにArcを過剰発現させたところ,そのシナプス刈り込み障害は通常レベルまで回復した.この結果から,登上線維からのシナプス伝達が減弱した結果としてプルキンエ細胞に流れ込むCa2+が減少し,Ca2+によって駆動される初期応答遺伝子Arcの発現量が不十分なことが,シナプス刈り込みの後期過程の異常を引き起こしていることが明らかとなった(図5).
9. C1ql1-BAI3の過剰発現によるシナプス刈り込みの促進
前述したCbln1と同じC1qファミリーに属する分泌分子であるC1ql1は,登上線維の起始核である延髄の下オリーブ核細胞に選択的に発現している42).最近,掛川らによりC1ql1が登上線維シナプスに集積することが示され,さらにC1ql1のノックアウトマウスが解析された結果,シナプス刈り込みへの関与が明らかとなった43).C1ql1ノックアウトマウスでは,プルキンエ細胞を支配する登上線維間で入力強度の差は生じるが,強い登上線維による入力が生後7日以降より強化されなかった.また過剰登上線維シナプスの刈り込みも生後7日の前期過程から障害がみられ,生後13日以降で顕著に障害された(図4F).さらに登上線維において出生直後からC1ql1を過剰発現させた結果,C1ql1が過剰発現したすべての登上線維においてシナプス入力の強度が増大し,弱い登上線維の除去が促進された.これにより,C1ql1は発現するすべての登上線維シナプスを強化し,さらには強い登上線維を残して弱い登上線維を除去する機能を持つことが明らかにされた.またC1ql1のシグナルをプルキンエ細胞で受け取る受容体も明らかにされた.BAI3はC1ql1と結合することが報告されている膜タンパク質である44).掛川らがBAI3のプルキンエ細胞特異的ノックアウトマウスを作製して解析したところ,強い登上線維による入力が強化されず,シナプス刈り込みも障害されるというC1ql1ノックアウトマウスと同様の異常が観察された.さらに,C1ql1に結合できない変異型BAI3と野生型BAI3をそれぞれプルキンエ細胞特異的BAI3ノックアウトマウスの小脳に発現させた.その結果,野生型BAI3はプルキンエ細胞特異的なBAI3ノックアウトマウスでみられた登上線維シナプスの異常を完全に回復させたが,変異型BAI3は回復させることができなかった.これらの結果から,プルキンエ細胞のBAI3受容体は登上線維からのC1ql1を受け取り,その結合が発達期小脳における登上線維シナプスの刈り込みを制御することが示された.さらに成熟マウスにおいて,登上線維でC1ql1を,プルキンエ細胞でBAI3をそれぞれノックダウンしたところ,これらのマウスのプルキンエ細胞では,登上線維シナプス入力が大きく減弱していた.以上より,登上線維–プルキンエ細胞シナプスにおけるC1ql1–BAI3シグナルは,生後発達期のシナプス刈り込みだけでなく,成熟したマウスにおいて強い登上線維シナプス入力の維持に重要な役割を担うことが明らかになった(図5).
本稿では,小脳の登上線維–プルキンエ細胞シナプスの生後発達について,これまでの研究の成果を概説したが,あらためてまとめると以下のようになる(図4,図5参照).マウスでは,生後7日までは,プルキンエ細胞は平均7本の登上線維から細胞体にシナプス入力を受けている.生後3日ではこれら複数の登上線維入力の強さはほぼ同じであるが,生後7日までにその中の1本だけが登上線維–プルキンエ細胞の同期的活動とP/Q VDCCを介するCa2+上昇に依存して選択的に強化される.その後,強くなった1本の登上線維だけが樹状突起にシナプス領域を拡大し,生涯にわたってプルキンエ細胞を支配するようになる.この単一登上線維の選択的強化,樹状突起移行,その後の維持には,IGF-1, Sema3A–PlxnA4経路,C1ql1–BAI3経路などの複数のシグナル経路が必要であると考えられる.一方,強くなった1本の登上線維以外の弱い登上線維のシナプスは,生後7日から17日ごろの間にプルキンエ細胞の細胞体から除去されるが,これは,平行線維シナプス形成とは無関係に起こる前期過程(生後11日ごろまで)と,平行線維シナプス形成に依存する後期過程(生後12日ごろ以降)に分かれる.前期過程にはP/Q VDCCを介するCa2+上昇が必須であり,C1ql1–BAI3が関与する.後期過程には,mGluR1–Gq–PLCβ4–PKCγのカスケードとその下流で働くSema7A–PlxnC1/ItgB1の経路とP/Q VDCC–Ca2+上昇によって活性化されるArcの経路が必須である.Cbln1–GluD2は平行線維–プルキンエ細胞シナプスを安定化させ,後期過程が正常に起こることを保障している.前期過程と後期過程を通じて,P/Q VDCCによるCa2+流入は弱いシナプスの刈り込みを促進させるが,Ca2+流入があるレベルを超えると,むしろ弱いシナプスを維持する働きがあることがわかった.籠細胞からのGABA作動性抑制は生後10日以降,プルキンエ細胞の細胞体でP/Q VDCCによるその過剰なCa2+流入を抑制して細胞体からのシナプス除去を促進し,TARPγ-2はAMPA受容体の機能を補助してP/Q VDCCおよびArcを適切に活性させることで,こちらもシナプス除去を促進することが明らかになった.
しかし依然として,登上線維シナプスの生後発達には謎が多く残されている.たとえば,強化される1本の登上線維の選択や樹状突起への移行メカニズム,P/Q VDCCのCa2+シグナルの下流因子やPKCγのリン酸化ターゲットなどの詳細な分子メカニズムはまだ明らかにされていない(図5).また小脳にはAldolase C/Zebrin IIやPLCβ4の発現パターンに代表される帯状構造があることが古くから知られているが32, 45),その構造の形成とシナプス刈り込みとの関連が示唆されているものの46),いまだ詳細な分子メカニズムは不明である.
一方,分子メカニズムだけでなく,登上線維シナプスの刈り込みの生理学的意義もまだはっきりわかっていない.P/Q VDCCやPKCγの変異はヒトやマウスにおいて,運動失調を引き起こすことが知られている47–50).また,BAI3のプルキンエ細胞特異的ノックアウトマウスでも視運動性反応において運動学習障害が認められている43).しかし多くの場合,登上線維のシナプス刈り込みに加えて平行線維における長期抑圧にも異常がみられており,シナプス刈り込みが運動障害にどれほど寄与しているかわかっていない.このように生後発達期の小脳における登上線維のシナプス刈り込みは,動物個体の発達や脳機能に重要であることに疑いはないが,その分子メカニズムや生理学的意義さらには疾患との関係を明らかにするためには,今後のさらなる研究が必要である.
謝辞Acknowledgments
本稿で紹介した研究は,主に東京大学大学院医学系研究科神経生理学分野で行われたものですが,多くの研究者との共同研究によって得られた成果です.また,C1ql1–BAI3についての最新の研究は,慶応義塾大学医学部柚崎通介教授および掛川渉講師らによってなされました.この場を借りて,引用させていただきました先生方と共同研究者の方々,ならびに日夜研究し続けている研究員・技術スタッフ・学生の皆様に厚く御礼申し上げます.
引用文献References
1) Hua, J.Y. & Smith, S.J. (2004) Nat. Neurosci., 7, 327–332.
2) Kano, M. & Hashimoto, K. (2009) Curr. Opin. Neurobiol., 19, 154–161.
3) Lichtman, J.W. & Colman, H. (2000) Neuron, 25, 269–278.
4) Riccomagno, M.M. & Kolodkin, A.L. (2015) Annu. Rev. Cell Dev. Biol., 31, 779–805.
5) Huttenlocher, P.R., de Courten, C., Garey, L.J., & Van der Loos, H. (1982) Neurosci. Lett., 33, 247–252.
6) Penzes, P., Cahill, M.E., Jones, K.A., VanLeeuwen, J.E., & Woolfrey, K.M. (2011) Nat. Neurosci., 14, 285–293.
7) Crepel, F., Mariani, J., & Delhaye-Bouchaud, N. (1976) J. Neurobiol., 7, 567–578.
8) Armengol, J.A. & Sotelo, C. (1991) Brain Res. Dev. Brain Res., 64, 95–114.
9) Hashimoto, K. & Kano, M. (2003) Neuron, 38, 785–796.
10) Hashimoto, K., Ichikawa, R., Kitamura, K., Watanabe, M., & Kano, M. (2009) Neuron, 63, 106–118.
11) Hashimoto, K., Yoshida, T., Sakimura, K., Mishina, M., Watanabe, M., & Kano, M. (2009) Neuroscience, 162, 601–611.
12) Hashimoto, K. & Kano, M. (2013) Cell. Mol. Life Sci., 70, 4667–4680.
13) Kashiwabuchi, N., Ikeda, K., Araki, K., Hirano, T., Shibuki, K., Takayama, C., Inoue, Y., Kutsuwada, T., Yagi, T., Kang, Y., Aizawa, S., & Mishina, M. (1995) Cell, 81, 245–252.
14) Kurihara, H., Hashimoto, K., Kano, M., Takayama, C., Sakimura, K., Mishina, M., Inoue, Y., & Watanabe, M. (1997) J. Neurosci., 17, 9613–9623.
15) Miyazaki, T., Hashimoto, K., Shin, H.S., Kano, M., & Watanabe, M. (2004) J. Neurosci., 24, 1734–1743.
16) Hashimoto, K., Tsujita, M., Miyazaki, T., Kitamura, K., Yamazaki, M., Shin, H.S., Watanabe, M., Sakimura, K., & Kano, M. (2011) Proc. Natl. Acad. Sci. USA, 108, 9987–9992.
17) Kakizawa, S., Yamada, K., Iino, M., Watanabe, M., & Kano, M. (2003) Eur. J. Neurosci., 17, 545–554.
18) Bosman, L.W., Hartmann, J., Barski, J.J., Lepier, A., Noll-Hussong, M., Reichardt, L.F., & Konnerth, A. (2006) Brain Cell Biol., 35, 87–101.
19) Johnson, E.M., Craig, E.T., & Yeh, H.H. (2007) J. Physiol., 582, 629–646.
20) Bosman, L.W., Takechi, H., Hartmann, J., Eilers, J., & Konnerth, A. (2008) J. Neurosci., 28, 798–807.
21) Ohtsuki, G. & Hirano, T. (2008) Eur. J. Neurosci., 28, 2393–2400.
22) Kawamura, Y., Nakayama, H., Hashimoto, K., Sakimura, K., Kitamura, K., & Kano, M. (2013) Nat. Commun., 4, 2732.
23) Uemura, T., Lee, S.J., Yasumura, M., Takeuchi, T., Yoshida, T., Ra, M., Taguchi, R., Sakimura, K., & Mishina, M. (2010) Cell, 141, 1068–1079.
24) Matsuda, K., Miura, E., Miyazaki, T., Kakegawa, W., Emi, K., Narumi, S., Fukazawa, Y., Ito-Ishida, A., Kondo, T., Shigemoto, R., Watanabe, M., & Yuzaki, M. (2010) Science, 328, 363–368.
25) Hashimoto, K., Ichikawa, R., Takechi, H., Inoue, Y., Aiba, A., Sakimura, K., Mishina, M., Hashikawa, T., Konnerth, A., Watanabe, M., & Kano, M. (2001) J. Neurosci., 21, 9701–9712.
26) Ichikawa, R., Miyazaki, T., Kano, M., Hashikawa, T., Tatsumi, H., Sakimura, K., Mishina, M., Inoue, Y., & Watanabe, M. (2002) J. Neurosci., 22, 8487–8503.
27) Hashizume, M., Miyazaki, T., Sakimura, K., Watanabe, M., Kitamura, K., & Kano, M. (2013) Front. Neural Circuits, 7, 130.
28) Hirai, H., Pang, Z., Bao, D., Miyazaki, T., Li, L., Miura, E., Parris, J., Rong, Y., Watanabe, M., Yuzaki, M., & Morgan, J.I. (2005) Nat. Neurosci., 8, 1534–1541.
29) Kano, M., Hashimoto, K., Kurihara, H., Watanabe, M., Inoue, Y., Aiba, A., & Tonegawa, S. (1997) Neuron, 18, 71–79.
30) Kano, M., Hashimoto, K., Chen, C., Abeliovich, A., Aiba, A., Kurihara, H., Watanabe, M., Inoue, Y., & Tonegawa, S. (1995) Cell, 83, 1223–1231.
31) Offermanns, S., Hashimoto, K., Watanabe, M., Sun, W., Kurihara, H., Thompson, R.F., Inoue, Y., Kano, M., & Simon, M.I. (1997) Proc. Natl. Acad. Sci. USA, 94, 14089–14094.
32) Kano, M., Hashimoto, K., Watanabe, M., Kurihara, H., Offermanns, S., Jiang, H., Wu, Y., Jun, K., Shin, H.S., Inoue, Y., Simon, M.I., & Wu, D. (1998) Proc. Natl. Acad. Sci. USA, 95, 15724–15729.
33) De Zeeuw, C.I., Hansel, C., Bian, F., Koekkoek, S.K., van Alphen, A.M., Linden, D.J., & Oberdick, J. (1998) Neuron, 20, 495–508.
34) Kakizawa, S., Yamasaki, M., Watanabe, M., & Kano, M. (2000) J. Neurosci., 20, 4954–4961.
35) Nakayama, H., Miyazaki, T., Kitamura, K., Hashimoto, K., Yanagawa, Y., Obata, K., Sakimura, K., Watanabe, M., & Kano, M. (2012) Neuron, 74, 384–396.
36) Lorenzetto, E., Caselli, L., Feng, G., Yuan, W., Nerbonne, J.M., Sanes, J.R., & Buffelli, M. (2009) Proc. Natl. Acad. Sci. USA, 106, 16475–16480.
37) Okuno, H., Akashi, K., Ishii, Y., Yagishita-Kyo, N., Suzuki, K., Nonaka, M., Kawashima, T., Fujii, H., Takemoto-Kimura, S., Abe, M., Natsume, R., Chowdhury, S., Sakimura, K., Worley, P.F., & Bito, H. (2012) Cell, 149, 886–898.
38) Mikuni, T., Uesaka, N., Okuno, H., Hirai, H., Deisseroth, K., Bito, H., & Kano, M. (2013) Neuron, 78, 1024–1035.
39) Uesaka, N., Mikuni, T., Hashimoto, K., Hirai, H., Sakimura, K., & Kano, M. (2012) J. Neurosci., 32, 11657–11670.
40) Uesaka, N., Uchigashima, M., Mikuni, T., Nakazawa, T., Nakao, H., Hirai, H., Aiba, A., Watanabe, M., & Kano, M. (2014) Science, 344, 1020–1023.
41) Kawata, S., Miyazaki, T., Yamazaki, M., Mikuni, T., Yamasaki, M., Hashimoto, K., Watanabe, M., Sakimura, K., & Kano, M. (2014) Cell Reports, 8, 1119–1129.
42) Iijima, T., Miura, E., Watanabe, M., & Yuzaki, M. (2010) Eur. J. Neurosci., 31, 1606–1615.
43) Kakegawa, W., Mitakidis, N., Miura, E., Abe, M., Matsuda, K., Takeo, Y.H., Kohda, K., Motohashi, J., Takahashi, A., Nagao, S., Muramatsu, S., Watanabe, M., Sakimura, K., Aricescu, A.R., & Yuzaki, M. (2015) Neuron, 85, 316–329.
44) Bolliger, M.F., Martinelli, D.C., & Sudhof, T.C. (2011) Proc. Natl. Acad. Sci. USA, 108, 2534–2539.
45) Brochu, G., Maler, L., & Hawkes, R. (1990) J. Comp. Neurol., 291, 538–552.
46) Miyazaki, T., Yamasaki, M., Hashimoto, K., Yamazaki, M., Abe, M., Usui, H., Kano, M., Sakimura, K., & Watanabe, M. (2012) J. Neurosci., 32, 1311–1328.
47) Chen, C., Kano, M., Abeliovich, A., Chen, L., Bao, S., Kim, J.J., Hashimoto, K., Thompson, R.F., & Tonegawa, S. (1995) Cell, 83, 1233–1242.
48) Chen, D.H., Brkanac, Z., Verlinde, C.L., Tan, X.J., Bylenok, L., Nochlin, D., Matsushita, M., Lipe, H., Wolff, J., Fernandez, M., Cimino, P.J., Bird, T.D., & Raskind, W.H. (2003) Am. J. Hum. Genet., 72, 839–849.
49) Jen, J.C., Graves, T.D., Hess, E.J., Hanna, M.G., Griggs, R.C., & Baloh, R.W., CINCH investigators (2007) Brain, 130, 2484–2493.
50) Pietrobon, D. (2010) Pflugers Arch., 460, 375–393.
著者紹介Author Profile
 渡邉 貴樹(わたなべ たかき)
渡邉 貴樹(わたなべ たかき)東京大学大学院医学系研究科神経生理学分野特任助教.博士(理学).
略歴2007年名古屋大学理学部卒業.09年同大学院理学研究科DC1, 日本学術振興会特別研究員.14年同博士課程修了.同年東京大学大学院医学系研究科神経生理学分野特任研究員.15年より現職.
研究テーマと抱負脳の生後発達とその分子基盤に興味を持ち研究しています.電気生理学・分子生物学的手法を中心に最先端の技術を取り入れつつ開発もしながら,シナプス刈り込みの分子基盤とその機能的な意味の全貌を解明していきたい.
趣味クラシック・ジャズピアノ,映画鑑賞.
 上阪 直史(うえさか なおふみ)
上阪 直史(うえさか なおふみ)東京大学大学院医学系研究科神経生理学分野助教.博士(理学).
略歴2001年大阪大学基礎工学部卒業.06年同大学院博士課程修了.同年大阪大学大学院医学系研究科特任研究員.10年より現職.
研究テーマと抱負脳の発達と進化に興味があります.神経科学の分野で貢献できるよう自分自身の研究を深めたいと思っております.
趣味スイーツを食べること.
 狩野 方伸(かのう まさのぶ)
狩野 方伸(かのう まさのぶ)東京大学大学院医学系研究科神経生理学分野教授.医学博士.
略歴1982年東京医科歯科大学医学部卒業,86年3月東京大学大学院医学系研究科博士課程修了,同年自治医科大学助手,90年講師,95年理化学研究所チームリーダー,98年金沢大学医学部教授,2005年大阪大学大学院医学系研究科教授,07年より現職.
研究テーマと抱負生後発達期に過剰なシナプスが刈り込まれて機能的な神経回路が作られる仕組みと,シナプス可塑性及び伝達調節の仕組みを研究しています.
ウェブサイトhttp://plaza.umin.ac.jp/~neurophy/Kano_Lab_j/Top_j.html
趣味名所旧跡めぐり.