Nrf2(NF-E2-related factor 2)は生体の恒常性維持に重要な転写因子である.Nrf2の主な標的遺伝子は最近の網羅的遺伝子発現解析やクロマチン免疫沈降法により明らかにされてきているが,その産物には解毒代謝や抗酸化などに働く酵素,また,重要な代謝系の酵素などが含まれている1).通常の環境ストレスがない条件下では,Nrf2は迅速にユビキチン化されて,プロテアソームにより分解されてしまうために,Nrf2タンパク質は細胞内に低いレベルでしか存在せず,その転写活性は低く抑えられている2).
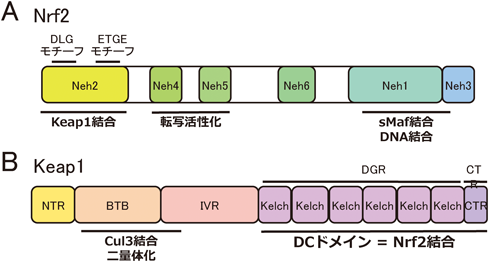
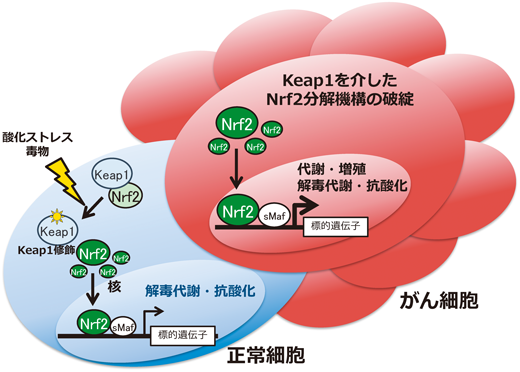
Keap1(Kelch-like ECH-associated protein 1)はNrf2のユビキチン化に関わるユビキチンE3リガーゼCullin 3(Cul3)のアダプター分子であるが3),そのC末端のDCドメインはNrf2タンパク質のN末端にある二つの結合モチーフ(DLGとETGEモチーフ)と直接結合している4).細胞が内外から毒物(親電子性分子のことが多い)や酸化ストレスに曝露されたとき,Keap1分子内の反応性の高いシステイン残基がそれらを検知する.すなわち,親電子性毒物や活性酸素はKeap1のシステイン残基のチオール基を酸化修飾するが,これによりKeap1とNrf2の結合が障害されて,Nrf2のユビキチン化反応が減弱する5).その結果,Nrf2の分解速度が低下し,安定化したNrf2が核に蓄積する.すなわち,核内におけるNrf2活性は,Nrf2タンパク質の恒常的分解からの脱抑制の形で誘導制御されている5).
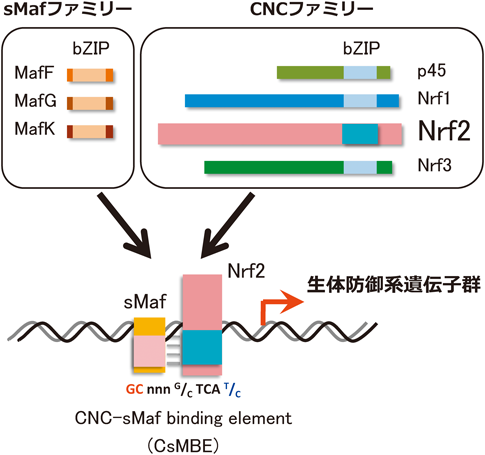
核内において,Nrf2は小Maf群因子とヘテロ二量体を形成し,抗酸化剤応答配列(antioxidant responsive element:ARE)6)または親電子性物質応答配列(electrophile responsive element:EpRE)7)に結合して,標的遺伝子の転写を活性化する.これらの配列は,従来,個別に記載されていたので混乱があったが,最近,CsMBE(CNC–sMaf binding element)として統一しようという提案もなされている8).Nrf2の活性化剤には,ジエチルマレイン酸のような実験試薬や,臨床薬として実用化されているジメチルフマル酸などが知られているが9),いずれもKeap1のシステイン残基を化学的に修飾することによってNrf2を活性化する(表1).なお,Nrf2の活性化は一過性であり,ストレスや誘導剤による標的遺伝子の活性化は次第に定常状態へ戻る.このように,Keap1–Nrf2システムは生体防御に働く制御系として発見され,その作用メカニズムが分子レベルで詳細に解析されてきた10).また,Nrf2の標的遺伝子産物には,解毒や酸化ストレス応答に働くものが多く含まれるため,本システムは創薬の視点からも注目されている.
表1 Nrf2活性化剤| 分 類 | 化合物名 | 略名/商品名/由来 |
|---|
| 実験試薬 | ジエチルマレイン酸 | DEM |
| tert-ブチルヒドロキノン | tBHQ |
| オルティプラッツ | |
| CDDO-イミダゾール | CDDO-Im |
| 食品添加物(酸化防止剤) | ブチルヒドロキシアニソール | BHA |
| 植物性サプリメント | スルフォラファン | ブロッコリースプラウト |
| クルクミン | ウコン |
| カルノシン酸 | ローズマリー |
| 臨床薬(多発性硬化症) | ジメチルフマル酸 | Tecfidera® |
| 臨床薬(糖尿病性腎臓病;開発中) | バルドキソロンメチル | RTA402/CDDO-Me |
表2 Keap1–Nrf2研究の年表| 年 | 内 容 | 発表者 |
|---|
| 1994, 1995 | Nrf2の発見 | Moiら11),Itohら12) |
| 1996, 1997 | Nrf2欠失マウスの作出 | Chanら15),Itohら16) |
| 1999 | Keap1の発見 | Itohら2) |
| 2001 | 老齢雌性Nrf2欠失マウスにおける自己免疫性腎炎 | Yohら18) |
| 2003 | Keap1欠失マウスの作出 | Wakabayashiら23) |
| 2003, 2004 | Neh2ドメインを介したKeap1によるNrf2タンパク質の分解 | Itohら36),Kobayashiら3) |
| 2004 | ヒトNRF2遺伝子多型 | Yamamotoら52) |
| 2004 | Nrf2欠失マウス切歯における鉄輸送の破綻 | Yanagawaら19) |
| 2006 | Keap1システイン残基のストレスセンサー | Kobayashiら5) |
| 2006 | 条件つきKeap1欠失マウスの作出 | Okawaら24) |
| 2006 | がん細胞におけるKeap1変異の発見 | Padmanabhanら28),Singhら29) |
| 2006, 2007 | Keap1–Nrf2結合におけるHinge–Latchモデル | Tongら4, 53, 54) |
| 2010 | Keap1ノックダウンマウスの同定 | Taguchiら25) |
| 2010 | オートファジーの破綻によるNrf2活性化 | Komatsuら38) |
| 2010 | Keap1タンパク質の結晶構造解析 | Oguraら41) |
| 2011 | 条件つきNrf2欠失マウスの作出 | Houら27) |
| 2011 | 条件つきKeap1欠失マウスの作出 | Kongら26) |
| 2012 | オートファジーによるKeap1タンパク質の分解 | Taguchiら39) |
| 2012, 2013 | Neh6ドメインを介したGSK3/βTrCPによるNrf2タンパク質の分解 | Radaら42),Chowdhryら43) |
| 2014 | Nrf2欠失マウスにおける門脈体循環シャント | Skokoら20) |
| 2016 | Nrf2欠失ラットの作出 | Taguchiら46),Priestleyら47) |
赤血球系転写因子NF-E2の同定の試みから,同転写因子がp45-sMafのヘテロ二量体であることが解明されたが,この転写因子は新しい塩基性領域・ロイシンジッパー(bZip)型のCNC–sMaf転写因子群の存在を実証したものであった.CNC因子群の新たなメンバーの探索研究から,ヒトNrf2がMoiら11)により,また,トリECHがItohら12)により同定されたが,両者はオルソログであり,その後は哺乳動物の本因子タンパク質はNRF2(ヒト),または,Nrf2(マウス・ラットなど)と呼称されてきた(表2).なお,CNC因子群として,他にNrf113)とNrf314)も同定されたが,解毒代謝の盛んな臓器・組織ではNrf2の発現が誘導的に非常に高くなるので,生体防御遺伝子群の発現制御にはNrf2が働くと予想された.
Nrf2の発見後すぐに,Nrf2欠失マウスの作出と解析が,米国のKanら15)と筆者の所属する本邦の山本ら16)により報告されたが,前者の解析はNrf2の血液系における機能解析に注力したものであった.すなわち,同じCNCファミリーに属するNF-E2のp45サブユニットの欠失マウスは血小板欠失による出血傾向を示すことから17),Nrf2欠失のマウス造血に対する影響に興味が持たれたが,Nrf2欠失マウスは正常に出生し,血球系細胞の分化と増殖に異常はみられなかった15).一方,後者の解析はNF-E2結合配列とARE/EpREの相同性に着目して,Nrf2の解毒代謝遺伝子群に対する制御を解析したものであった(図1).その結果,Nrf2欠失マウスは通常の飼育環境では致死に至るような顕著な表現型を示さないが,多くの親電子性毒物や酸化ストレスの曝露に対してきわめて脆弱であることが判明した.さらに,抗酸化剤であるブチルヒドロキシアニソール(BHA)によるNAD(P)Hキノン酸化還元酵素1(NAD(P)H quinone oxidoreductase 1:Nqo1)やグルタチオンS-転移酵素(glutathione S-transferase:GST)の遺伝子発現がNrf2依存的であることが,Nrf2欠失マウスを用いて実証された16).
その後のNrf2欠失マウスの解析は,本分野に多くの重要な知見をもたらした.マウスにおけるNrf2の欠失は若齢で重篤な表現型を示さなかったが,雌性Nrf2欠失マウスは自己免疫性腎炎を発症し,60週齢以降に死亡した18).その他にNrf2欠失マウスの表現型として知られているのは,切歯の白色化19),門脈体循環シャント20),肝重量比の低下21)である.これらについて,以下に簡潔に説明する.YanagawaらはNrf2欠失マウスの切歯が野生型マウスと比較して白色化していることを発見した19).げっ歯類の切歯は鉄の沈着により黄色く着色するが,Nrf2はフェリチンの遺伝子発現を制御しており,Nrf2欠失マウスの切歯は鉄輸送の破綻により鉄沈着が減少し,それにより白色化する.同時に,酸に対する耐性も失うので,う歯が進行する.一方,2014年にNrf2欠失マウスにおける門脈体循環シャントが発見された20).門脈体循環シャントとは,門脈と下大静脈の間に形成される短絡血管のことで,門脈血が肝臓を経ずに静脈血として直接体循環に流入してしまう.イヌなどに往々にしてみられ,尿石症などの症状を示すことから,獣医学の分野でよく知られる表現型である.Nrf2と同様に,解毒代謝に関わる遺伝子の発現を制御する転写因子である多環芳香族炭化水素受容体(aryl hydrocarbon receptor:AhR)欠失マウスでも門脈体循環シャントが報告されているが22),なぜNrf2やAhRの欠失により門脈体循環シャントが生じるのか,門脈体循環シャント形成の分子メカニズムや解毒代謝酵素とシャント形成の相関は明らかでない.また,Nrf2欠失マウスの肝重量比が小さいことが報告されているが,その主たる原因は明らかでなく,肝細胞の増殖能の低下がその一因ではないかと考えられている.
Nrf2の発見から5年後の1999年に,Nrf2の制御に重要なKeap1が発見された2).図2AにヒトNrf2とトリECH(Nrf2のトリオルソログ)の保存性比較から提唱されたドメイン構造を示す2).Keap1は,酵母ツーハイブリット法によりNrf2のNeh2ドメインに結合するタンパク質として同定された2).
ところで,Keap1欠失マウスを作出する際には,同マウスはNrf2欠失マウスと相反して,生体防御遺伝子群の恒常的な活性化により親電子性毒物や酸化ストレスに抵抗性を示す非常に健康なスーパーマウスであることが予想された.しかしながら,まったく予期せぬことにKeap1欠失マウスは食道や前胃に重篤な過角化という表現型を示し,生後1か月以内に全例死亡してしまった.一方,Keap1::Nrf2二重欠失マウスでは,Keap1単独欠失マウスのような食道の過角化はみられなかったので,ケラチン層の過剰形成はKeap1欠失によるNrf2の活性化に依存することが実証された.本事象のため,Keap1欠失マウスは当初企画したような解析に供することはできなかった23).
そこで,条件つきKeap1欠失(Keap1flox)マウスが作出された24).本マウスとKeratin 5-Creマウスとの交配により作出した基底細胞特異的Keap1欠失(Keap1-K5)マウスは,全身性Keap1欠失マウスと同様に食道・前胃における過角化を示したので25),上述の扁平上皮におけるNrf2異常活性化によるKeap1欠失マウスの死亡がさらに検証された.一方,筆者らはこのKeap1floxマウスは全身性にKeap1発現が低下しているKeap1遺伝子ノックダウン(Keap1-KD)マウスであることに気がついた.本Keap1-KDマウスでは,食道・前胃に多少の角化がみられるが症状が軽度のために致死性は回避されている.他方で,このKeap1-KDマウスは,段階的なKeap1発現量を示すマウスの作出を可能にした点が特筆される.この点を活用して,筆者は段階的なNrf2活性の制御により,生体防御系酵素群の発現とその副作用が段階的に制御されていることを明らかにした.その後,Biswalらにより新たなノックダウンにならない条件つきKeap1欠失マウスが26),また,Piらにより条件つきNrf2欠失マウス27)も作出されている.なお,筆者らが作出したKeap1ノックダウンマウスはKeap1flox-Aマウス,BiswalらのものはKeap1flox-Bマウスと呼称されて,多くの研究に活用されている.
4. がん細胞における転写因子Nrf2の異常活性化
筆者がNrf2研究に本格的に着手し始めたのは2006年であったが,同年にはNrf2研究においていくつかの大きなブレイクスルーがあった.一つは,ヒトがん細胞におけるKEAP1遺伝子の体細胞変異によるNRF2異常活性化の発見である(図3)28, 29).Ohtaら30)により発見された肺がん細胞におけるKEAP1体細胞変異は,KEAP1分子のNRF2結合ドメインに集積していた(図2B).この変異はKEAP1とNRF2の結合を減弱し,NRF2分解を停止させるために,NRF2の活性化を招来する.その後,肺がんのみならず,さまざまな組織由来のがん細胞でKEAP1変異やNRF2遺伝子変異が次々と報告されている10).
特筆すべきことは,NRF2遺伝子の体細胞変異はKEAP1との結合に重要なNeh2ドメインに存在するDLGモチーフとETGEモチーフに集積しているという点である10).さらには,遺伝子変異のみならず,KEAP1プロモーター領域のメチル化によるサイレンシング31),p62タンパク質の異常蓄積32),フマル酸のようながん代謝物によるKEAP1システイン残基の修飾33)など,がん細胞においてNRF2の異常活性化を導く分子メカニズムが明らかとなった.これらはすべて,NRF2とKEAP1の結合モデルに基づいており,NRF2がユビキチン化されてプロテアソームにより分解されるという分子基盤に立脚している.
NRF2が異常活性化した代表的ながん細胞株は,ヒト肺がんA549細胞である.A549細胞はKEAP1遺伝子のKelchモチーフからなるdouble-glycine-rich(DGR)領域にG→Tの点変異があり,これはKEAP1タンパク質にG333Cのアミノ酸変異を与える34).NRF2 siRNAによりNRF2の発現を減弱させると,A549細胞の増殖は有意に遅延する35).冒頭に記したように,NRF2は解毒代謝や抗酸化に働く遺伝子の発現を制御する転写因子である.すなわち,がん細胞におけるNRF2の異常な活性化は恒常的な解毒や抗酸化に関わる生体防御遺伝子群の発現を亢進し,がん細胞に抗がん剤による化学療法や放射線治療による抵抗性を賦与する.
これらのメカニズムに加えて,筆者らはNRF2の標的遺伝子産物として新たにペントースリン酸経路やグルタミン代謝に関わる代謝酵素群を同定した.すなわち,NRF2が活性化しているがん細胞においては,解毒や抗酸化に関わる生体防御遺伝子群の発現亢進のみならず,NRF2活性化ががん細胞の代謝リプログラミングに働くことで,その悪性化に働いていることが示された35).
5. 二つのタンパク質分解系による統合的なNrf2の制御メカニズム
1)Keap1タンパク質の分解機構:オートファジー
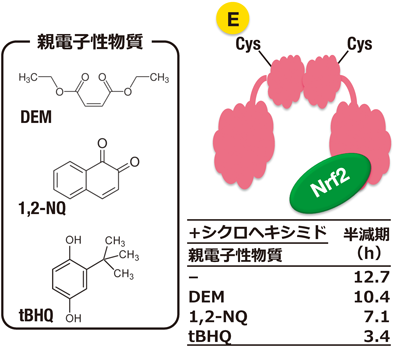
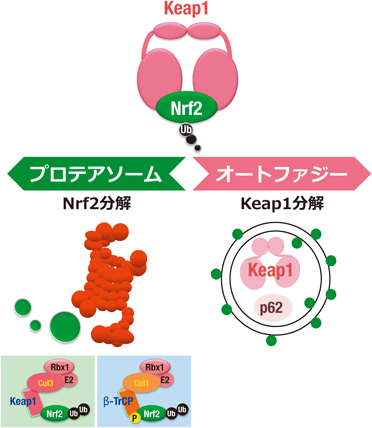
Nrf2がユビキチンで標識されてプロアソーム系で選択的に分解されるタンパク質であることはよく知られているが3, 36),Keap1タンパク質の分解系は明らかではなかった.Keap1タンパク質の分解はプロテアソーム系に依存しないという否定的な報告があったが37),その詳細は不明であった.筆者は,オートファジー研究に関して小松雅明教授(新潟大学)と共同研究する機会を得たので,肝臓特異的Atg7遺伝子欠失(Atg7-Alb)マウスの解析を実施し,タンパク質分解機構であるオートファジーの機能が破綻するとp62タンパク質が異常蓄積してNrf2が恒常的に活性化する,という新たなNrf2活性化メカニズムの発見に参画した38).この際に,不思議なことにAtg7-Albマウスの肝臓においてKeap1タンパク質量が増加していることを見いだした.この観察の以前には,Keap1タンパク質量が劇的に変化する現象はなかったので,Keap1タンパク質の分解にオートファジーが関与するのではないかと考えた.Keap1タンパク質の半減期は約12時間で,Nrf2(半減期18分)と比べるとその分解はずっと遅い.そこで,Keap1タンパク質のシステイン残基を修飾する親電子性物質3種をそれぞれHepG2細胞に曝露して,Keap1タンパク質の半減期を調べたところ,程度に差はあるものの,いずれの場合にも親電子性物質を曝露するとKeap1の分解が速まることが明らかとなった(図4)39).すなわち,これらの結果から,p62に結合したKeap1はともにタンパク質凝集体に巻き込まれて分解されることが明らかとなった.
この結果は,プロテアソームとオートファジーという二つの細胞内タンパク質分解系の両方を利用して,Keap1–Nrf2ストレス応答系の精密な制御が行われていることを示すものである(図5).興味深いことに,筆者らの論文公刊の翌年に,Keap1タンパク質がオートファジーで分解されるという分子メカニズムは,栄養飢餓の際のセストリンによるNrf2の活性化メカニズムの基盤を形成していることが,韓国のRheeらにより明らかにされた40).
2)Nrf2タンパク質の分解機構:ユビキチン-プロテアソーム
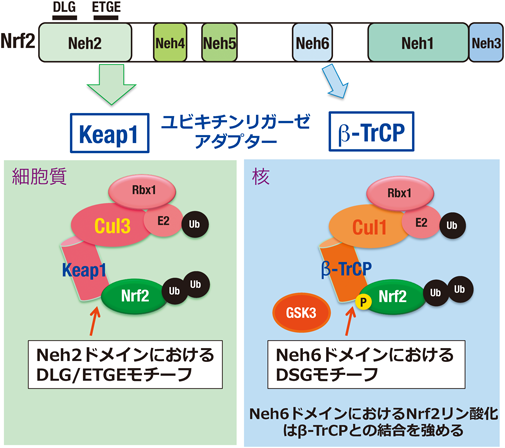
Nrf2がユビキチンにより標識されてプロテアソーム系で選択的に分解されるタンパク質であることは2003年と2004年に発表された論文で明らかにされたが,ユビキチン化の分子機構とNrf2誘導についての研究がその後積み重ねられている3, 36).Keap1はユビキチンE3リガーゼのアダプター分子としてCul3-Rbx型ユビキチンE3リガーゼ複合体を形成する.1分子のNrf2は分子内のNeh2ドメインに存在するDLGモチーフおよびETGEモチーフを介してKeap1ホモ二量体(Keap1としては2分子)と結合している.このKeap1–Nrf2構造は,電子顕微鏡によって可視化され,柄の末端でつながった2個のさくらんぼ(cherry-bob)の形であることが示されている41).
一方,2012年42)と2013年43)にスペインのCuadradoらは,Nrf2のNeh6ドメインにおけるDSGモチーフがグリコーゲン合成酵素キナーゼ3(glycogen synthase kinase 3:GSK3)によってリン酸化されること,さらに,リン酸化されるとNeh6モチーフとβ-TrCP(β-transducin repeat-containing protein)との結合が強まることを報告した.β-TrCPもKeap1と同様にユビキチンE3リガーゼを形成するが,サブユニットとしてCul1と結合してCul1–Rbx型ユビキチンE3リガーゼ複合体を形成する.すなわち,GSK3によるDSGモチーフのリン酸化は,Nrf2の分解を促進する(図6).
GSK3はPI3K(phosphoinositide 3-kinase)–Akt(v-Akt murine thymoma viral oncogene)経路の下流に位置し,リン酸化によってその活性が制御されている.GSK3は通常条件下で活性化しているが,一方,Aktによりリン酸化されると不活性型になる.そのため,Ptenが欠失したがん細胞においては,Aktリン酸化に引き続くGSK3リン酸化により,GSK3は不活性化となり,Nrf2はリン酸化されずに分解を免れて活性化している.しかしながら,肝臓特異的Pten欠失マウスを解析したところ,若齢ではNrf2の活性化はみられなかった44).したがって,β-TrCP–GSK3経路よりもKeap1–Cul3経路によるNrf2活性化の方が優位にあると考えられた.がん細胞におけるNrf2の活性化メカニズムを考える際には,Keap1に依存しないPI3K–Akt経路の破綻によるNrf2の異常活性化も考慮する必要がある.
6. Nrf2欠失ラットの作出:アフラトキシンの解毒代謝とNrf2
ところで,Keap1–Nrf2システムの研究は,Nrf2欠失マウスとKeap1欠失マウスの利用により飛躍的に発展してきた経緯がある.一方で,マウスでは再現できない実験モデルもあり,実験動物としてマウスに限界があることも否定できない.たとえば,ラットは古くから実験動物として利用されてきたので,特に毒性実験などのデータが蓄積している.しかし,ラット胚性幹細胞(ES細胞)は2008年に樹立が報告されたものの45),長い間安定した利用がかなわなかったので,マウスに比べて遺伝子改変動物ラットの作出は遅れていた.
ゲノム編集技術は,ES細胞を利用せずに遺伝子改変動物を作出することを可能にした画期的な技術である.ゲノム編集技術は,ZFN(zinc finger nuclease),TALEN(transcription activator-like effector nuclease),または,CRISPR/Cas(clustered regularly interspaced short palindromic repeats/ CRISPR-associated protein)などのヌクレアーゼを利用して,標的遺伝子を改変する画期的な手法である.そこで,このゲノム編集技術による遺伝子改変ラットの作出が注目されることになった.
筆者もこの点に着目して,数年前からNrf2欠失ラットの作出に取り組んできた.真下知士博士(大阪大学)にご協力いただいて,F344系統のラットにZFNを用いたゲノム編集を行い,成功裏にNrf2欠失ラットを作出し,本年に論文として公刊した46).なお,Lombardら47)によりWister系統に対してTALENを用いたゲノム編集を実施して作出したNrf2欠失ラットも,本年報告されている.筆者らの作出したNrf2欠失ラットを解析したところ,Nrf2欠失マウスと同様に,胎生致死にならずに正常に出生した.Nrf2欠失マウスでみられた切歯の白色化と肝重量比の低下も観察された.しかし,Nrf2欠失マウスでみられた門脈体循環シャントはラットでは明確でなく,この表現型はマウスでもラットでも系統に依存するものであると考えられる.
ラットでもNrf2活性化剤であるCDDO-ImによってNrf2が活性化するが,同剤はNrf2欠失ラットでは効果を発揮しないので,同剤はNrf2依存的に抗酸化や解毒代謝に関わる遺伝子の発現を亢進させるものと理解される.筆者は,長く毒性学で利用されてきたF344系統ラットの利点を活用して,マウスでは再現できない実験である,発がん物質アフラトキシンB1(AFB1)による肝毒性に対する生体防御におけるNrf2の貢献を調べた.AFB1はさまざまな食品に繁殖するAspergillus属菌が産生する毒物である.アフラトキシンは10数種の関連物質の総称であるが,特にAFB1の毒性が高く,肝細胞がんの原因となる.日本でもピーナッツなどの輸入食品中からアフラトキシンがたびたび検出されている.
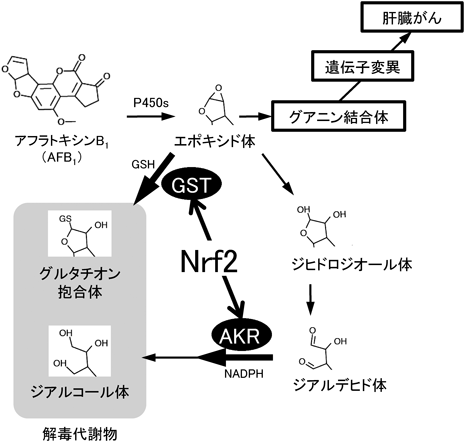
ところで,マウスではAFB1の毒性が再現できないのはなぜだろうか? それは,マウス特有のGSTの高い発現にあると考えられる48).すなわち,AFB1の解毒反応では,まず初めにシトクロムP450によりエポキシド体を形成し,これがDNAのグアニン残基に結合して遺伝子変異を誘導する.一方,エポキシド体は,GSTとアルド-ケト還元酵素(aldo-keto reductase:AKR)によりさらに解毒される.簡潔に紹介すると,GSTはエポキシド体にグルタチオン(glutathione:GSH)を結合する反応を触媒して,グルタチオン抱合体の形成を促進する.一方,エポキシド体は加水分解によりジヒドロジオール体を介してジアルデヒド体を形成する経路を進むこともあるが,AKRはジアルデヒド体からジアルコール体への反応を触媒する.このような解毒反応を俯瞰すると,マウスはGSTの発現レベルが高いため,エポキシド体はグルタチオン抱合体として迅速に解毒排泄され,それによる遺伝子変異が起こりにくいので,AFB1の毒性が再現されないと考えられる(図7).
なお,Nrf2はGSTA3およびGSTA5, AKR7A2およびAKR7A3の遺伝子発現を制御して,この解毒反応を促進している.筆者は,Kensler教授(ピッツバーグ大学)との共同研究により,野生型ラットでは致死量でないAFB1をNrf2欠失ラットに反復投与すると,約半数が致死となることを見いだした46).また,この際にNrf2欠失ラットでは解毒代謝物の生成が有意に少なかった.したがって,Nrf2欠失ラットはAFB1解毒代謝が機能せず,AFB1の肝毒性に対して脆弱性を示すことが実証された.
アフラトキシン肝発がんモデルと同じように,マウスでは再現できず,ラットが実験動物として利用される実験モデルとして,1977年に報告されたSolt-Farber肝発がんモデルがある49).本モデルは,発がん物質であるジエチルニトロサミン(DEN)と2-アセチルアミノフルオレン(2AAF)に部分肝切除を組み合わせて肝臓がんを誘導する.早期に出現する少数の細胞からなるGST陽性の病巣(foci)は,非常に増殖が盛んで,前がん小結節(preneoplastic nodule)から肝臓がんを形成する.ちょうど40年前に確立された本モデルは,1997年に報告されたラット肝中期発がん性試験法(伊東法)の礎になっており,2AAFの代わりに被験物質を投与することで発がん性の有無を調べることができる50).最近,イタリアのColumbanoらは,Solt-FarberモデルにおけるGST陽性病巣には早期からNrf2遺伝子に体細胞変異が出現することを発見した51).筆者らの作出したNrf2欠失ラットを用いれば,Solt-Farberモデルに対するNrf2の貢献を検討することが可能になるものと思われる.また,発がん性を示す化学物質の約60%は肝臓を標的にしているといわれているので,上述のラット肝中期発がん性試験法を用いて,種々の化学物質による肝発がん過程におけるNrf2の関与を示すことができるものと期待される.
謝辞Acknowledgments
本研究を遂行するにあたり,東北大学大学院医学系研究科医化学分野山本雅之教授にご指導いただきました.ここに深甚なる謝意を表します.また,研究室の皆様や,共同研究の先生方に大変お世話になりました.本研究室においてKeap1–Nrf2研究に携わることができたことに深謝申し上げます.
引用文献References
1) Hirotsu, Y., Katsuoka, F., Funayama, R., Nagashima, T., Nishida, Y., Nakayama, K., Engel, J.D., & Yamamoto, M. (2012) Nucleic Acids Res., 40, 10228–10239.
2) Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J.D., & Yamamoto, M. (1999) Genes Dev., 13, 76–86.
3) Kobayashi, A., Kang, M.I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., Igarashi, K., & Yamamoto, M. (2004) Mol. Cell. Biol., 24, 7130–7139.
4) Tong, K.I., Katoh, Y., Kusunoki, H., Itoh, K., Tanaka, T., & Yamamoto, M. (2006) Mol. Cell. Biol., 26, 2887–2900.
5) Kobayashi, A., Kang, M.I., Watai, Y., Tong, K.I., Shibata, T., Uchida, K., & Yamamoto, M. (2006) Mol. Cell. Biol., 26, 221–229.
6) Rushmore, T.H. & Pickett, C.B. (1990) J. Biol. Chem., 265, 14648–14653.
7) Friling, R.S., Bensimon, A., Tichauer, Y., & Daniel, V. (1990) Proc. Natl. Acad. Sci. USA, 87, 6258–6262.
8) Otsuki, A., Suzuki, M., Katsuoka, F., Tsuchida, K., Suda, H., Morita, M., Shimizu, R., & Yamamoto, M. (2016) Free Radic. Biol. Med., 91, 45–57.
9) Suzuki, T., Motohashi, H., & Yamamoto, M. (2013) Trends Pharmacol. Sci., 34, 340–346.
10) Taguchi, K., Motohashi, H., & Yamamoto, M. (2011) Genes Cells, 16, 123–140.
11) Moi, P., Chan, K., Asunis, I., Cao, A., & Kan, Y.W. (1994) Proc. Natl. Acad. Sci. USA, 91, 9926–9930.
12) Itoh, K., Igarashi, K., Hayashi, N., Nishizawa, M., & Yamamoto, M. (1995) Mol. Cell. Biol., 15, 4184–4193.
13) Chan, J.Y., Han, X.L., & Kan, Y.W. (1993) Proc. Natl. Acad. Sci. USA, 90, 11371–11375.
14) Kobayashi, A., Ito, E., Toki, T., Kogame, K., Takahashi, S., Igarashi, K., Hayashi, N., & Yamamoto, M. (1999) J. Biol. Chem., 274, 6443–6452.
15) Chan, K., Lu, R., Chang, J.C., & Kan, Y.W. (1996) Proc. Natl. Acad. Sci. USA, 93, 13943–13948.
16) Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., Oyake, T., Hayashi, N., Satoh, K., Hatayama, I., Yamamoto, M., & Nabeshima, Y. (1997) Biochem. Biophys. Res. Commun., 236, 313–322.
17) Shivdasani, R.A., Rosenblatt, M.F., Zucker-Franklin, D., Jackson, C.W., Hunt, P., Saris, C.J., & Orkin, S.H. (1995) Cell, 81, 695–704.
18) Yoh, K., Itoh, K., Enomoto, A., Hirayama, A., Yamaguchi, N., Kobayashi, M., Morito, N., Koyama, A., Yamamoto, M., & Takahashi, S. (2001) Kidney Int., 60, 1343–1353.
19) Yanagawa, T., Itoh, K., Uwayama, J., Shibata, Y., Yamaguchi, A., Sano, T., Ishii, T., Yoshida, H., & Yamamoto, M. (2004) Genes Cells, 9, 641–651.
20) Skoko, J.J., Wakabayashi, N., Noda, K., Kimura, S., Tobita, K., Shigemura, N., Tsujita, T., Yamamoto, M., & Kensler, T.W. (2014) Toxicol. Sci., 141, 112–119.
21) Zhang, Y.K., Wu, K.C., & Klaassen, C.D. (2013) PLoS ONE, 8, e59122.
22) Harrill, J.A., Hukkanen, R.R., Lawson, M., Martin, G., Gilger, B., Soldatow, V., LeCluyse, E.L., Budinsky, R.A., Rowlands, J.C., & Thomas, R.S. (2013) Toxicol. Appl. Pharmacol., 272, 503–518.
23) Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., Imakado, S., Kotsuji, T., Otsuka, F., Roop, D.R., Harada, T., Engel, J.D., & Yamamoto, M. (2003) Nat. Genet., 35, 238–245.
24) Okawa, H., Motohashi, H., Kobayashi, A., Aburatani, H., Kensler, T.W., & Yamamoto, M. (2006) Biochem. Biophys. Res. Commun., 339, 79–88.
25) Taguchi, K., Maher, J.M., Suzuki, T., Kawatani, Y., Motohashi, H., & Yamamoto, M. (2010) Mol. Cell. Biol., 30, 3016–3026.
26) Kong, X., Thimmulappa, R., Craciun, F., Harvey, C., Singh, A., Kombairaju, P., Reddy, S.P., Remick, D., & Biswal, S. (2011) Am. J. Respir. Crit. Care Med., 184, 928–938.
27) Hou, Y., Xue, P., Bai, Y., Liu, D., Woods, C.G., Yarborough, K., Fu, J., Zhang, Q., Sun, G., Collins, S., Chan, J.Y., Yamamoto, M., Andersen, M.E., & Pi, J. (2012) Free Radic. Biol. Med., 52, 462–472.
28) Padmanabhan, B., Tong, K.I., Ohta, T., Nakamura, Y., Scharlock, M., Ohtsuji, M., Kang, M.I., Kobayashi, A., Yokoyama, S., & Yamamoto, M. (2006) Mol. Cell, 21, 689–700.
29) Singh, A., Misra, V., Thimmulappa, R.K., Lee, H., Ames, S., Hoque, M.O., Herman, J.G., Baylin, S.B., Sidransky, D., Gabrielson, E., Brock, M.V., & Biswal, S. (2006) PLoS Med., 3, e420.
30) Ohta, T., Iijima, K., Miyamoto, M., Nakahara, I., Tanaka, H., Ohtsuji, M., Suzuki, T., Kobayashi, A., Yokota, J., Sakiyama, T., Shibata, T., Yamamoto, M., & Hirohashi, S. (2008) Cancer Res., 68, 1303–1309.
31) Wang, R., An, J., Ji, F., Jiao, H., Sun, H., & Zhou, D. (2008) Biochem. Biophys. Res. Commun., 373, 151–154.
32) Inami, Y., Waguri, S., Sakamoto, A., Kouno, T., Nakada, K., Hino, O., Watanabe, S., Ando, J., Iwadate, M., Yamamoto, M., Lee, M.S., Tanaka, K., & Komatsu, M. (2011) J. Cell Biol., 193, 275–284.
33) Adam, J., Hatipoglu, E., O’Flaherty, L., Ternette, N., Sahgal, N., Lockstone, H., Baban, D., Nye, E., Stamp, G.W., Wolhuter, K., Stevens, M., Fischer, R., Carmeliet, P., Maxwell, P.H., Pugh, C.W., Frizzell, N., Soga, T., Kessler, B.M., El-Bahrawy, M., Ratcliffe, P.J., & Pollard, P.J. (2011) Cancer Cell, 20, 524–537.
34) Taguchi, K., Shimada, M., Fujii, S., Sumi, D., Pan, X., Yamano, S., Nishiyama, T., Hiratsuka, A., Yamamoto, M., Cho, A.K., Froines, J.R., & Kumagai, Y. (2008) Free Radic. Biol. Med., 44, 1645–1655.
35) Mitsuishi, Y., Taguchi, K., Kawatani, Y., Shibata, T., Nukiwa, T., Aburatani, H., Yamamoto, M., & Motohashi, H. (2012) Cancer Cell, 22, 66–79.
36) Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., O’Connor, T., & Yamamoto, M. (2003) Genes Cells, 8, 379–391.
37) Zhang, D.D., Lo, S.C., Sun, Z., Habib, G.M., Lieberman, M.W., & Hannink, M. (2005) J. Biol. Chem., 280, 30091–30099.
38) Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., Sou, Y.S., Ueno, I., Sakamoto, A., Tong, K.I., Kim, M., Nishito, Y., Iemura, S., Natsume, T., Ueno, T., Kominami, E., Motohashi, H., Tanaka, K., & Yamamoto, M. (2010) Nat. Cell Biol., 12, 213–223.
39) Taguchi, K., Fujikawa, N., Komatsu, M., Ishii, T., Unno, M., Akaike, T., Motohashi, H., & Yamamoto, M. (2012) Proc. Natl. Acad. Sci. USA, 109, 13561–13566.
40) Bae, S.H., Sung, S.H., Oh, S.Y., Lim, J.M., Lee, S.K., Park, Y.N., Lee, H.E., Kang, D., & Rhee, S.G. (2013) Cell Metab., 17, 73–84.
41) Ogura, T., Tong, K.I., Mio, K., Maruyama, Y., Kurokawa, H., Sato, C., & Yamamoto, M. (2010) Proc. Natl. Acad. Sci. USA, 107, 2842–2847.
42) Rada, P., Rojo, A.I., Evrard-Todeschi, N., Innamorato, N.G., Cotte, A., Jaworski, T., Tobon-Velasco, J.C., Devijver, H., Garcia-Mayoral, M.F., Van Leuven, F., Hayes, J.D., Bertho, G., & Cuadrado, A. (2012) Mol. Cell. Biol., 32, 3486–3499.
43) Chowdhry, S., Zhang, Y., McMahon, M., Sutherland, C., Cuadrado, A., & Hayes, J.D. (2013) Oncogene, 32, 3765–3781.
44) Taguchi, K., Hirano, I., Itoh, T., Tanaka, M., Miyajima, A., Suzuki, A., Motohashi, H., & Yamamoto, M. (2014) Mol. Cell. Biol., 34, 900–913.
45) Li, P., Tong, C., Mehrian-Shai, R., Jia, L., Wu, N., Yan, Y., Maxson, R.E., Schulze, E.N., Song, H., Hsieh, C.L., Pera, M.F., & Ying, Q.L. (2008) Cell, 135, 1299–1310.
46) Taguchi, K., Takaku, M., Egner, P.A., Morita, M., Kaneko, T., Mashimo, T., Kensler, T.W., & Yamamoto, M. (2016) Toxicol. Sci., 152, 40–52.
47) Priestley, J.R., Kautenburg, K.E., Casati, M.C., Endres, B.T., Geurts, A.M., & Lombard, J.H. (2016) Am. J. Physiol. Heart Circ. Physiol., 310, H478–H487.
48) Wild, C.P. & Turner, P.C. (2002) Mutagenesis, 17, 471–481.
49) Solt, D.B., Medline, A., & Farber, E. (1977) Am. J. Pathol., 88, 595–618.
50) Ito, N., Hasegawa, R., Imaida, K., Hirose, M., Shirai, T., Tamano, S., & Hagiwara, A. (1997) J. Toxicol. Pathol., 10, 1–11.
51) Zavattari, P., Perra, A., Menegon, S., Kowalik, M.A., Petrelli, A., Angioni, M.M., Follenzi, A., Quagliata, L., Ledda-Columbano, G.M., Terracciano, L., Giordano, S., & Columbano, A. (2015) Hepatology, 62, 851–862.
52) Yamamoto, T., Yoh, K., Kobayashi, A., Ishii, Y., Kure, S., Koyama, A., Sakamoto, T., Sekizawa, K., Motohashi, H., & Yamamoto, M. (2004) Biochem. Biophys. Res. Commun., 321, 72–79.
53) Tong, K.I., Kobayashi, A., Katsuoka, F., & Yamamoto, M. (2006) Biol. Chem., 387, 1311–1320.
54) Tong, K.I., Padmanabhan, B., Kobayashi, A., Shang, C., Hirotsu, Y., Yokoyama, S., & Yamamoto, M. (2007) Mol. Cell. Biol., 27, 7511–7521.
著者紹介Author Profile
 田口 恵子(たぐち けいこ)
田口 恵子(たぐち けいこ)東北大学大学院医学系研究科医化学分野助教.博士(医学).
略歴2000年東京理科大学理工学部卒業.02年筑波大学大学院環境科学研究科修士課程修了.06年筑波大学大学院人間総合科学研究科社会環境医学専攻修了.同年科学技術振興機構ERATO研究員.08年より現職.
研究テーマと抱負Keap1–Nrf2システムと生体応答.
ウェブサイトhttp://www.dmbc.med.tohoku.ac.jp/official/index.html