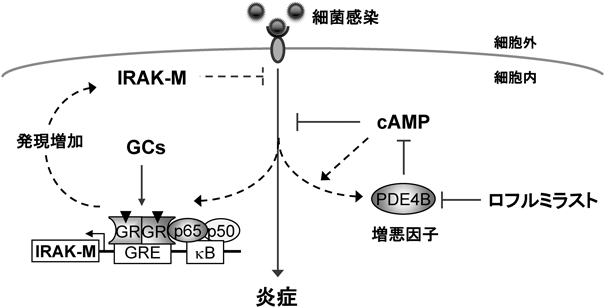
抗炎症薬による細胞内シグナルフィードバックの制御Regulation of feedback signaling by anti-inflammatory drugs
関西学院大学理工学部生命医化学科Department of Biomedical Chemistry, Graduate School of Science and Technology, Kwansei Gakuin University ◇ 〒669–1337 兵庫県三田市学園2–1 ◇ 2–1 Gakuen, Sanda, Hyogo 669–1337, Japan
発行日:2016年10月25日Published: October 25, 2016