1. ミエリンを作る細胞と機能,それらに関する最近の知見
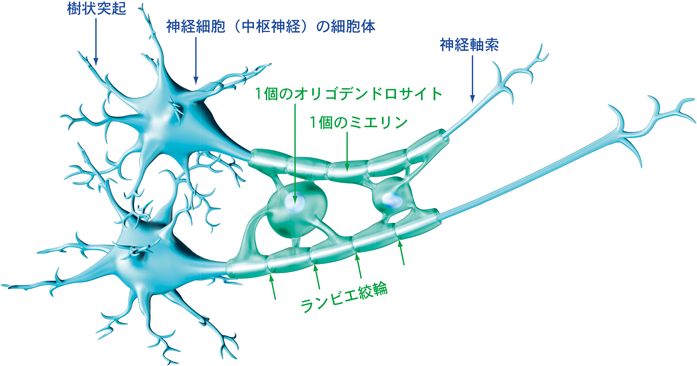
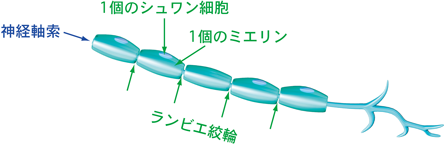
髄鞘(ミエリン)は,神経細胞の周りにある鞘(さや)のような構造体である.その輪切り断片をみると,真ん中に神経の線維を持つ年輪のようなかたちをしている1, 2).普通の神経細胞は,情報の入力を担う樹状突起,細胞核がある細胞体,情報の出力を担う神経軸索の3種類の細胞領域に大別されるが,ミエリンは神経軸索の周りにのみ作られる.そして,それは脳や脊髄からなる中枢神経系の神経細胞と,からだの至るところに張り巡らされている感覚神経などの末梢神経系の神経細胞に存在する.末梢神経系では,シュワングリア細胞(シュワン細胞)が,中枢神経系では,希突起膠細胞(オリゴデンドログリア細胞,オリゴデンドロサイト)がミエリン膜を作り,それを伸長させることで,神経軸索の周りに複層構造が形成され,ミエリンができる(図1および図2).すなわち,両神経系に存在する細胞系譜のまったく異なった,特異的なグリア細胞の細胞膜が形態的に分化してミエリンを作るのである3–12).
ミエリンの第一の機能は,跳躍伝導を誘導する重要な担い手であることである.神経軸索の周りを巻くミエリンの存在部位と次のミエリンの存在部位の間に,イオンチャネルが豊富に存在するわずかな隙間のような構造(ランビエ絞輪)がある(図1および図2).跳躍伝導とはイオン電流がこの隙間と次の隙間へと‘跳躍’することである.跳躍伝導によって神経細胞の電気伝導速度が,100倍またはそれ以上に上昇する.しかし,この跳躍伝導を担うミエリンの長さは,均一ではない.それが偶然なのか,必然的なものなのか不明である.また,シュワン細胞が作るミエリンは500 µmを超えるものも多く,一般的に長い.一方,オリゴデンドロサイトが作るミエリンはそこまでの長さを持たない.これは,末梢神経系と中枢神経系の神経軸索の長さの相違(前者は末梢組織まで到達する必要性があるため平均的にミエリンの長さが長く,後者は短い)に反映されているのかもしれない.さらに,最近,中枢神経系においてミエリンの存在が不均一であることが報告された13).つまり,1本の神経軸索上にはミエリン存在部位と非存在部位があり,神経軸索上を流れるイオン電流の速度が部分的に一定でないことを意味している.また,ランビエ絞輪に存在するイオンチャネルは,ミエリン形成前に配置されている場合もあるようだ14).すなわち,ミエリンとその周辺構造が‘固い’イメージを持つ構造体であるというよりはむしろ,必要性に応じて柔軟に,その部分構造や形成過程の一部を変化させることができるものであるようだ.
もう一つの重要なミエリンの機能は,神経軸索の保護である.これは一般的なグリア細胞の主要機能と類似している.ミエリンによって外部環境から神経軸索が構造的に保護(物理的な側面)され,かつ,神経軸索側に栄養が供給(生物的な側面)される.
神経軸索の周りには何十層ものミエリン膜が巻かれる(図3および図4).このミエリンの層の数は,神経線維の輪切り断面を電子顕微鏡で観察すると容易に判別できる.厚いミエリンは末梢神経系にみられることが多いが,これは末梢組織の空間が脳や脊髄の内部空間よりも大きいため,この空間的要素がミエリン膜の層数を決めると考えると理にかなう.確かに,末梢神経系では100層以上の厚さを持つミエリンが少なくない.末梢神経系では神経線維の外側は血管など神経系以外の組織であり,ミエリンが外部環境との境目となる.そのため,一定の厚さのミエリンが構造的に必要になる.ハンセン病は主にシュワン細胞のなかにマイコバクテリウムが寄生する感染症である.感染によりシュワン細胞を前駆細胞のような状態に戻してしまう15).その結果,シュワン細胞が本来の機能を発揮できず,神経線維と末梢組織との境界が消失し,ハンセン病のさまざまな症状が現れると考えられている.ミエリンが神経線維と末梢組織の間に壁を作り,それを保護する役割を持つ重要性がわかる.
一方,ミエリンは血管を通して運搬された栄養分子を異化し,それを神経軸索側に供給する役割も有する.一般的に,シュワン細胞に,このような機能が備わっていることは知られていた.オリゴデンドロサイトにも,この機能があることが,数年前に証明された16).オリゴデンドロサイトは,他の細胞と同様に,エネルギー源の一つとしてグルコースを取り込み,解糖系でそれをピルビン酸にし,TCAサイクル以下の代謝経路でATPを作る.特に,オリゴデンドロサイトがミエリン膜を作るときは大量のATPが消費されることが知られている.このとき,オリゴデンドロサイトの解糖系はピルビン酸ばかりではなく,ピルビン酸の次の産物である乳酸も大量に生産する.この乳酸は神経細胞とのネットワークを介して,神経軸索に運ばれる.これが神経細胞の栄養源になる.
オリゴデンドロサイトのTCAサイクルには,エネルギー生産への関与ばかりではなく,ミエリンの脂質成分を作るという重要な役割があるが,これは他の総説を参考にしていただきたい7, 8).
以上のようにミエリンの構造や機能に関する研究だけを抜粋しても,ここ数年,重要な研究成果が次々に発表された.それは,現在に至ってもなお,ミエリンやミエリンを持つ神経細胞(有髄神経)の研究が未開拓分野であることを示している.さらに,末梢神経系に関する研究報告が比較的多くなってきている.それは末梢神経系の主要な構成細胞がシュワン細胞と神経細胞の2種類であるため,中枢神経系の研究に比べて研究を進めやすいからかもしれない.本稿では「末梢神経系のミエリン発生に関する分子メカニズム」を中心に,まず現在までのミエリン発生に関する知見を述べ,次に新しいプレーヤー分子であるサイトヘジン(Cytohesin)の役割を紹介したい.
2. 転写因子と受容体によるミエリン形成細胞の制御
シュワン細胞とオリゴデンドロサイトのミエリン発生のスタートポイントは,それぞれの前駆細胞がミエリン形成能を持つ細胞に分化するところから始まる.マウスやラットなどのげっ歯類の末梢神経では,生後すぐにシュワン細胞がミエリンを形成するようになる.脳のオリゴデンドロサイトは,生後すぐの時期では前駆細胞のままであり,数日後に分化し,ミエリン形成期に入る.広義のミエリン発生期は,それぞれの前駆細胞が増殖し遊走する胎生中後期からの時期を含めることもあるが,これは最近では一般的ではないようだ5–12).
さて,ミエリン発生のスタートポイントに関して,どのような実験的根拠に基づき,ミエリン形成能を有する細胞であると判別するのだろうか.シュワン細胞では,発生時間軸に沿って発現変化する転写因子や表面抗原などの分化マーカーがあまり同定されていない.つまり,シュワン細胞前駆細胞(Schwann cell precursor)と,その前後の細胞の状態を区別できるマーカーがない.そのため,免疫組織化学的方法を用いて,神経組織内でのシュワン細胞の分化状態を明らかにすることは容易なことではない.厳密に各細胞のミエリン発生のスタートポイントやその分化状態を決めるのは難しい.一方,オリゴデンドロサイトにおいては複数のマーカーが明らかにされており,市販の分化マーカー抗体での二重染色も可能であるため,後述するように,神経組織内でオリゴデンドロサイトの分化状態を正確に把握できる.
また,シュワン細胞前駆細胞がシュワン細胞以外の細胞の前駆細胞でもあると考えられているため,シュワン細胞系譜に関する情報が複雑化している3, 4).たとえば,シュワン細胞と色素細胞が発生段階のある時期までは同一の細胞系譜であることはよく知られている.ごく最近,Dyachukら17)とEspinosa-Madinaら18)の研究グループは,それぞれ独立して,頭蓋部のシュワン細胞前駆細胞が,ホメホドメイン型転写因子Phox2bを発現する副交感神経細胞を作り出すことを明らかにした.シュワン細胞前駆細胞がどのような性質を持つ細胞であるのか,どのように定義すべきなのか,今後も議論されると思われる.
シュワン細胞前駆細胞も,他の細胞と同様に,非対称分裂を繰り返すことで,さまざまな細胞を作り出すと考えられている.そのため,シュワン細胞前駆細胞には細胞極性に関与する分子が多く発現している.興味深いことに,これらの分子の発現はミエリン形成時期まで続く.細胞極性制御に重要な分子であるPar3とp75低親和性神経栄養因子受容体がミエリン膜の先端に局在し,そのミエリン化にも関与していることが知られている19, 20).
1)転写因子
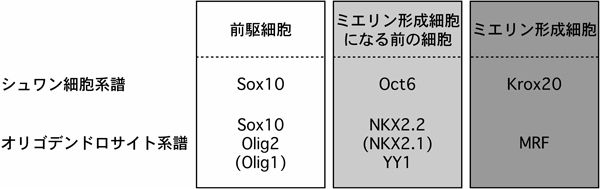
ミエリンを形成するシュワン細胞になるまで,いくつかの代表的な転写因子がその過程を制御することが知られている(図5).high mobility group(HMG)ボックス型Sox10→ホメホドメイン型Oct6(別名POU3F1またはSCIP)→ジンク・フィンガー型Krox20(別名Egr2)の順番に発現が誘導され,最終的にミエリン形成シュワン細胞に成熟すると考えられている21–24).特に,Sox10は胎生中期以降,シュワン細胞系譜の細胞を決める重要な発現マーカーである.それはシュワン細胞の発生過程においてSox10が継続的に発現しているからである.Sox10は生後に緩やかに発現減少するが,消失することはないようだ.
Sox10は神経系を中心に多様な役割を持つため,完全ノックアウトマウスは胎生後期で致死になる3, 4).そのため,シュワン細胞系列に特異的なコンディショナル・ノックアウトマウスが作製された.しかし,その結果も重篤で,未成熟なシュワン細胞が末梢神経組織内に蓄積し,神経変性症のような病理形態を示した.Sox10は,ミエリン形成不全を含む神経変性症であるPeripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome, and Hirschsprung disease(PCWH)症候群や,神経障害を含む多岐にわたる病態を呈するWaardenburg症候群の原因遺伝子として知られている.これらの疾患において既知のSox10変異は,そのほとんどがloss-of-function(機能欠失)型の変異であると考えられている.
Oct6やKrox20も胎生期から発現がみられ,それぞれのノックアウトマウスの解析から,Oct6は主としてミエリンを形成する細胞への分化に関与し,Krox20はミエリン膜の複層構造形成段階に関与することが明らかになっている.しかし,Sox10→Oct6→Krox20間の連携は,連続的ではなさそうである.たとえば,Sox10が発現上昇し,それがOct6プロモーター上のSox10結合配列部分に結合することで,生体内においてOct6が発現誘導されるかどうか明らかになっていない.Sox10のDNA結合配列であるACA ANN(NはAの可能性が高い)は転写因子のDNA認識配列としては比較的短く,多くの遺伝子のプロモーター上にあるため,Oct6の誘導には未同定の転写因子も多く関与していると推定される.また,Krox20がミエリン形成に必須な,すべての膜タンパク質を含む構造タンパク質や脂質代謝関連酵素群の発現を誘導するかというと,Krox20だけでは荷が重すぎるように感じる.Wegnerらが先導し,これらの転写因子の研究を促進させた21–24).しかし,シュワン細胞における未同定の転写因子も多くあると考えられる.
一方,オリゴデンドロサイトでは,経時的に発現変化する複数の転写因子が明らかにされている(図5).現在まで明らかにされたオリゴデンドロサイト転写因子の一部は連続的な発現誘導ループにあることがわかっている.しかし,すべての因子が連続的に発現誘導するか今後の解明を待たねばならない.また,Luらを中心にオリゴデンドロサイトの転写因子と直接的,間接的に相互作用するクロマチンのリモデリング因子に関する研究も精力的になされ(たとえば文献25),それらの全容解明が待たれる段階になってきている.
オリゴデンドロサイトにおいてもSox10は主要な転写因子の一つであるが,先述したPCWH症候群でみられる多様な病態から推定されるように,脳ではミエリン形成オリゴデンドロサイトへの発生過程に関する役割以外にも,さまざまな役割を持つことが知られている.そのため,Sox10よりも若干後期に発現誘導されるOlig2やOlig1がオリゴデンドロサイト系譜の細胞を決める発現マーカーとして用いられ,特にマーカーとしてのOlig2の特異性は高いと考えられている.Olig2とOlig1のどちらも,オリゴデンドロサイト前駆細胞から発現している重要な転写因子であることは確かである.
現在までに明らかにされているオリゴデンドロサイトの転写因子の種類は非常に多いので,代表的な3種類の転写因子を記載する.それらはホメホドメイン型NKX2.2(NKX2.1も必要であるとされる)およびジンク・フィンガー型Yin-Yang-1(YY1)→Myelin regulatory factor(MRF,別名MYRF)であり,この順に発現が誘導される.NKX2.2およびYY1とMRFは,それぞれ,シュワン細胞におけるOct6とKrox20のような働きを持つ転写因子であると解釈されている.MRFはユニークな転写因子であり,ホメホドメインなどの典型的なDNA結合領域を持たない26).これらのオリゴデンドロサイト転写因子を含むほとんどの転写因子は,主要なミエリン膜内外の構造タンパク質や脂質代謝関連酵素を発現誘導することが知られている.
2)細胞接着分子
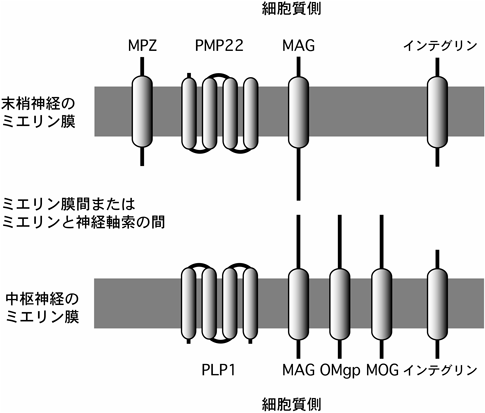
シュワン細胞に発現している主要な細胞接着分子はmyelin protein zero(MPZ,別名P0またはZero)と呼ばれ,ミエリン構成タンパク質の約半分を占め1回膜貫通型の糖タンパク質である(図6).糖鎖が付加されても実質的な分子量は30,000前後の小さなタンパク質である.ミエリン膜間において逆平行でホモフィリックな結合様式を示す.また,細胞接着分子としてミエリン膜間には22 kDa peripheral myelin protein(PMP22)という4回膜貫通構造を有する糖タンパク質が存在する.しかし,ミエリンタンパク質としての含有量は5%以下である.MPZと同様に,ミエリン膜間において逆平行でホモフィリックな結合様式を示す.MPZもPMP22も末梢神経でのミエリン膜とミエリン膜をつなぐ役割を持つタンパク質である.このことから,ミエリン膜間に存在する細胞接着分子は,同質の「細胞膜接着分子」と呼ばれた方が正確であるとされる9, 10).
ミエリンの複層構造には内側と外側があり,内側は神経軸索に面し,外側はベーサル・ラミナ(基底)に面している.シュワン細胞が作るミエリンの内側の主要な細胞接着分子はmyelin-associated glycoprotein(MAG)である.MAGはSiglec-4とも呼ばれ,神経軸索側の糖鎖に結合するレクチンとしての役割がある.また,多様な分子の受容体になることも知られており,多様な分子のリガンドになることもあるようだ.ミエリン複層構造の外側は細胞外基質に結合するα6タイプのインテグリンが発現している.インテグリンには複数のαとβサブユニットの分子種があり,それらがヘテロ二量体を形成することで多様なリガンドと結合する能力を得る.α6タイプのインテグリンもそうである.興味深いことに,ミエリンが形成される前,そのβサブユニットはβ1であるが,形成期にはβ4に変化する.α6β4インテグリンは,細胞外基質であるラミニンのさまざまなタイプと結合できるため,α6β1→α6β4のサブユニット変化はラミニンへの認識能の広がりを意味している.ミエリンタンパク質としての含有量は,MAGが約1%,α6インテグリンではそれ以下である9, 10).
一方,オリゴデンドロサイトが作るミエリン膜には,MPZのようなタンパク質は存在しないと考えられている.これに代わって,proteolipid protein 1(PLP1)と呼ばれるPMP22に類似した4回膜貫通構造を持ち,同様の機能を持つ分子がある.その含有量はオリゴデンドロサイトのミエリン構成タンパク質の約半分を占めている.
また,MAGや何種類かのインテグリンも発現していることが知られている.それぞれの含有量はシュワン細胞由来のミエリン膜と同程度である.他には,MAGと構造が類似したoligodendrocyte myelin glycoprotein(OMgp,別名OMG)やmyelin oligodendrocyte glycoprotein(MOG)などがある.これらの分子の主要な機能は細胞接着であると推定されているが,不明な点が多い.2000年代初頭にMAGやOMgpは,Nogoと同様に,神経軸索の伸長阻害に関与するオリゴデンドロサイト側のリガンドであることが明らかにされた27).ミエリン形成時の現象と神経軸索の伸長阻害との関係は複雑である.さらに,最近,MOGは神経成長因子(nerve growth factor:NGF)と結合することが示され,MOGに受容体機能があることが示唆された28).
ここまでいくつかの細胞接着分子を列挙したが,これらの分子やそれを裏打ちする構造タンパク質をそれぞれ単独でノックアウトしても,ほとんどの場合,ミエリンが消失するような強い効果は出現しない.それは転写因子のノックアウトの表現型と対照的である.細胞接着分子の場合,細胞接着分子間またはその裏打ち構造タンパク質間での相補性が強いか,未知のホモログが発現し,1分子をノックアウトしてもミエリンの複層構造が維持できるのかもしれない.
3)細胞増殖因子の受容体
Neuregulin 1(NRG1)は,神経細胞に発現し,シュワン細胞系譜細胞の生存維持からミエリン発生までを正に制御するグリア細胞の増殖因子として同定された.NRG1は上皮増殖因子(epidermal growth factor:EGF)ファミリーに属する因子である.オリゴデンドロサイトにおいても,NRG1はシュワン細胞系譜細胞における役割と類似した機能があることが判明している5, 6).
現在,NRG1には選択的スプライシングにより生成される15種類以上のバリアントがあることが知られている7, 8).ミエリン発生過程に関与するNRG1のバリアント(III型)は初め,細胞膜を貫通した状態で神経細胞上に提示され,その後,何らかの刺激に応じてBACE1やADAMファミリープロテアーゼで切られ,細胞膜から遊離することでシュワン細胞のミエリン化を制御する.NRG1は,シュワン細胞上で,EGF受容体ファミリーに属するErbB3とErbB2のヘテロ二量体に結合する.ErbB3とErbB2は細胞外ドメインがロイシンリッチおよびシステインリッチドメインから構成されるプロトタイプ型の細胞増殖因子受容体である.ErbB3はNRG1と結合するがチロシンキナーゼ活性はほとんどなく,逆に,ErbB2はNRG1とほとんど結合できないがチロシンキナーゼ活性を有している.したがって,NRG1はErbB3に結合し,ErbB2を介してシュワン細胞内に,そのシグナルが伝達される.
NRG1のいくつかのバリアントのコンディショナル・ノックアウトマウスも作製され,それらの解析から,主要なバリアントのノックアウトマウスは感覚神経のミエリン発生に重篤な障害を持っていることが示されている.一方,ErbB3とErbB2のコンディショナル・ノックアウトマウスの解析も進み,こちらはNRG1のノックアウトマウスの表現型とほぼ同じであった7, 8).
中枢神経におけるNRG1のノックアウトマウスの解析では,オリゴデンドロサイトにおいて表現型を示さなかった.しかし,オリゴデンドロサイトのNRG1受容体であるErbB4は,統合失調症や自閉症の関連遺伝子として注目を集めている29).一般的に,オリゴデンドロサイトでのミエリン発生に関与する増殖因子受容体のノックアウトマウスの表現形質は,一部の例外を除き,転写因子のそれに比べて弱い.それは,オリゴデンドロサイトが,外部環境に影響を受けにくい,連続的な転写因子の発現カスケードである「オリゴデンドロサイト・タイマー」と呼ばれる潜在的な分子カスケードを持ち,そのタイマーに従って連続的に前駆細胞から成熟したオリゴデンドロサイトへと分化するからであると考えられている30).一方,シュワン細胞には,そのようなタイマーがなく,外部環境の刺激がその後のシュワン細胞の運命を決定づけると考えられている3, 4).
ErbB3/2と類似したプロトタイプ型の構造を持つ細胞増殖因子受容体であるインスリン様増殖因子1(insulin-like growth factor 1:IGF1)受容体もシュワン細胞に発現している.IGF1受容体はミエリン化を正に制御することが証明されている31).また,そのリガンドであるIGF1も,主として神経細胞から放出されている11, 12, 32).IGF1はオリゴデンドロサイトにおいてもミエリン化の促進因子であることが知られている33, 34).
免疫グロブリン様ドメイン構造を持つ増殖因子受容体である血小板由来増殖因子(platelet-derived growth factor:PDGF)および線維芽細胞増殖因子(fibroblast growth factor:FGF)の受容体もシュワン細胞に発現し,ミエリン化において重要な役割を果たすことがin vitroで証明されている35).しかし,これらのリガンドと受容体には複数のファミリー分子があり,それそれがシュワン細胞やオリゴデンドロサイトおよびそれらの周辺細胞に発現している.たとえば,脳内では,PDGF受容体αはオリゴデンドロサイト前駆細胞に発現しているが,PDGF受容体βは脳血管の内皮細胞の外側を囲んでいるペリサイトに発現している36).それぞれの役割を証明するためには,in vitroにおける実験の他,コンディショナル・ノックアウトマウスを用いた詳細な解析が必要となるだろう.
4)Gタンパク質共役受容体
NRG1を多く含むとされる脳の抽出液とフォルスコリン(サイクリックAMPを上昇させる化合物)は,旧来,グリア細胞の培養に欠かせない培養液添加物であった.しかし,シュワン細胞のサイクリックAMPを上昇させる受容体が発見されたのは最近のことである.ゼブラフィッシュの遺伝学的解析から,それがGタンパク質共役受容体126(GPR126)であることが明らかにされた.ゼブラフィッシュにはミエリン形成グリア細胞という一群の細胞群があり,gpr126変異体では,その形成が不完全であった37).GPR126は哺乳動物でも同様の機能を持ち,リガンドとしてIV型コラーゲンやラミニン211が候補としてあげられている38, 39).GPR126はadhesion GPCR(またはGPR)という一群のGタンパク質共役受容体ファミリーに含まれ,長いN末端部分を持つ.一般的に,このタイプのGPCRはN末端部分が切られることで活性化されるため,その活性化に特異的なリガンドを必要としない.そのため,GPR126の活性化機構も同様のメカニズムである可能性があり,今後の研究が期待される.
GPR126はオリゴデンドロサイトには発現していないが,GPR56が発現している.しかし,オリゴデンドロサイトのGPR56の役割は,ミエリン化促進というGPR126の役割と異なっている.GPR56はオリゴデンドロサイト前駆細胞の増殖期に限定的に発現し,主に前駆細胞の増殖を制御し,ミエリン化には負に働くようだ40).他に,オリゴデンドロサイトにおいて,古典的なニューロペプチドやリン脂質およびエイコサノイドに対するGPCRやGPR17, GPR30, GPR37, GPR68など遺伝子単離時にはオーファン受容体とされていた受容体が,その発生に関与していることが報告されている40).しかし,これらのGPCRのなかでサイクリックAMPを特異的に上昇させる受容体はない.オリゴデンドロサイトの培養にもサイクリックAMPを上昇させる化合物を添加することが多いが,それは複数の因子の添加物の一部でしかなく,オリゴデンドロサイトに対する発生過程における細胞内サイクリックAMPの影響は限られているのかもしれない.
5)発生過程における受容体と転写因子の関係
シュワン細胞においては,細胞接着因子や増殖因子受容体と転写因子の発現は,相互関係にあると考えられている5–12).たとえば,Sox10をノックアウトするとErbB3やErbB2の受容体の発現が減少するが,逆に,ErbB3やErbB2をノックアウトしてもSox10の発現が減少する.受容体と転写因子の巧妙な相互作用の関係の上に,ミエリン発生が成立しているのかもしれない.一方,オリゴデンドロサイトでは受容体と転写因子の関係に関する報告は多くない.
1)サイトヘジンの分子種
サイトヘジン1(Cytohesin-1またはCyth1)は,90年代半ばに,ヒトJurkat細胞のcDNAライブラリーから酵母ツーハイブリッド法を用いてlymphocyte function-associated antigen-1(LFA-1)中のβ2インテグリン・サブユニットの細胞質領域に結合するタンパク質として同定された分子で,それが名称の由来の一部にもなっている41–43).全長のアミノ酸配列が明らかにされると,酵母の分泌経路に関与するSEC7と相同性を持つB2-1と同一の分子であることが判明した41)(本稿ではサイトヘジン1を,その統一名称として用いる).現在,哺乳動物にもSEC7と相同性が高い領域を持つ分子が複数あることが知られ,その相同性領域をSec7ドメインと呼んでいる(後述).また,ほぼ同時期に,マウスのサイトヘジン1のエキソン周辺配列もクローニングされ44),それが早期の遺伝子改変マウスの作製に役立った.
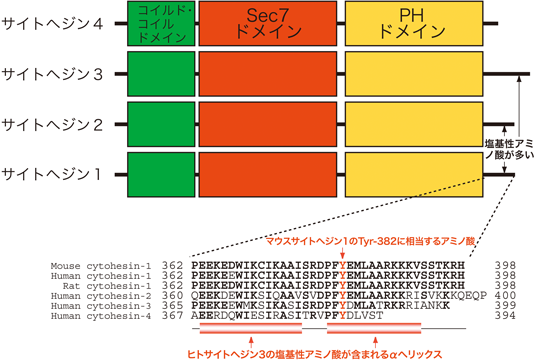
さて,サイトヘジン1はN末端側から隣接した3種類のドメインと特徴的なC末端配列から構成されている.それらはコイルド・コイル(coiled-coil)ドメイン,Sec7ドメイン,pleckstrin homology(PH)ドメイン,C末端の塩基性アミノ酸に富む配列である.サイトヘジン1は45~50 kDaの小さなタンパク質であるため,これらのドメイン以外に機能ドメインはないと考えられている.
コイルド・コイルドメインは6個おきにロイシンなどの分枝鎖アミノ酸が点在するモチーフから構成され,サイトヘジン1どうしまたは他のコイルド・コイルドメインを持つ分子との結合に関与している.
Sec7ドメインは低分子量GTP結合タンパク質のArfファミリーの交換因子(または活性化因子)活性を有する触媒ドメインで,サイトヘジン1の主要な機能を担う.低分子量GTP結合タンパク質はGDP型が不活性型でGTP型が活性化型である.通常は,シグナル依存的にGTP型になることでエフェクター分子にシグナルを伝達する.交換因子は,これをGDP型からGTP型へ交換する活性を持つ分子である.
Sec7ドメインに続くPHドメインはリン脂質に結合する.サイトヘジン1のPHドメインはイノシトールリン脂質に特異的に結合することが知られており,線維芽細胞ではイノシトールリン脂質シグナル依存的にサイトヘジン1が細胞膜にリクルートされる.最もC末端に位置する塩基性アミノ酸に富む配列は,PHドメインが細胞膜に結合するのを助ける役割があると考えられている.PHドメインとそれに続く領域と相互作用する分子は,サイトヘジン1の活性化に関与する分子であると推定される(後述).
ヒトのサイトヘジン1には3種類のスプライシングバリアントがあることが知られている.それらは,全長399アミノ酸を持つ最も長いもの,C末端部分で1アミノ酸の少ない398アミノ酸を持つもの,N末端部分が欠けている340アミノ酸である.
これらの研究と並行して,サイトヘジン1がファミリーを形成していることも明らかにされた.クローニング順に,サイトヘジン2(ヒト:401と400アミノ酸からなる2種類のアイソフォームがある),サイトヘジン3(ヒト:400アミノ酸),サイトヘジン4(ヒト:395とN末端部分が欠けている338アミノ酸からなる2種類のアイソフォームがある)と命名され,全ファミリーが明らかにされた45–49).
サイトヘジン1における399と398アミノ酸を持つアイソフォーム間,および,サイトヘジン2における401と400アミノ酸を持つアイソフォーム間の相違は,PHドメイン内の1アミノ酸である.このアイソフォーム間の性質の相違は詳細に調べられており,PHドメインのイノシトールリン脂質への結合特異性に影響することが明らかにされている.サイトヘジン1と3の長いアイソフォームの方のPHドメインは2種類のイノシトールリン脂質[PtdIns(4,5)P2とPtdIns(3,4,5)P3]に特異的に結合する.一方,それらよりも1アミノ酸少ないアイソフォームは1種類のイノシトールリン脂質[PtdIns(3,4,5)P3]に特異的に結合する50, 51).PHドメインのイノシトールリン脂質への選択性が,自然界にあるアイソフォーム間の1アミノ酸の相違で決定されることはきわめて珍しい.
サイトヘジンはArfファミリーを不活性型のGDP結合型から活性型のGTP結合型にする交換因子(guanine-nucleotide exchange factor)であるが,その特異性に関して統一的な見解はない.その理由は特異性検討時の実験方法にあるようだ.ある研究グループは触媒ドメインであるSec7ドメインのみを大腸菌で発現させ特異性を調べ,あるグループはSec7ドメインまたは全長をヒトやマウスの細胞に発現させ,それらの特異性を調べている.in vitroでは,どのサイトヘジンのSec7ドメインも,強弱の差こそあれ,Arf1とArf6の両方に対して活性を持つようだ.一方,シグナル依存的に活性化された全長のサイトヘジン(1または2)は,Arf1よりもむしろArf6を活性化するようである52).
Arfファミリーの交換因子は,サイトヘジン・ファミリー以外に11種類知られ,4種類のファミリーに分類されている53, 54).それらはADP-ribosylation factor guanine-nucleotide exchange factor 1~3(Arfgef1~3,別名BIG1~3),pleckstrin and Sec7 domain containing 1~4(PSD1~4,別名EFA6A/C/D/B),IQ motif and Sec7 domain 1~3(IQSEC1~3,別名BRAG2/1/3),F-box protein 8(FBXO8)と命名されており,Arfgefファミリーの分子だけがArf1に比較的高い特異性を持つ.Arfgef以外の交換因子はArf6に特異性を持つようだ.
これらの交換因子の共通の特徴は,すべてSec7ドメインを持つことである.しかし,それぞれの分子の長さはさまざまであり,それぞれ特有の機能ドメインを持つ.一方,Sec7ドメイン以外の触媒ドメインを持つArfファミリーの交換因子の存在も推定されている.その理由はArf1とArf6以外のArfファミリー分子も哺乳動物にあるからである.
ところで,サイトヘジン1から4は類似したドメイン構造を持つが,発現組織がそれぞれ異なる.サイトヘジン1は,脾臓などに多く,血球系や免疫系の細胞に発現している.サイトヘジン2と3は全臓器に発現が確認され,さまざまな細胞に発現している.各臓器において,サイトヘジン2の方が3よりやや発現が高い.ただし,脂肪細胞のように,サイトヘジン3が2に比べて豊富に発現している細胞もある.サイトヘジン4の発現パターンは1に類似しているが,詳細に調べられていない.
2)サイトヘジン結合タンパク質
当初,サイトヘジン1の同定の経緯から,サイトヘジン2, 3, 4も細胞接着分子の細胞質側に結合しているシグナル複合体に含まれる分子であることが推定されていた.サイトヘジン1に関しては,それがLFA-1(αLβ2インテグリン)であり,サイトヘジン1がLFA-1と結合することで,LFA-1のインテグリンとしての機能発現に役立っていることが明らかにされている55).サイトヘジン2や3もインテグリンと結合することが知られている56).2010年にCell誌に「サイトヘジン・ファミリーがErbB受容体ファミリーに結合することでErbB受容体変異によるがん化を促進する」と報告された.サイトヘジン・ファミリーがErbB受容体と機能的に相互作用をすることは,多くの研究グループで確認された.しかし,両者の直接的な会合の確認が難しく,2016年初めにこの報告は訂正された.
サイトヘジン2が免疫細胞のアダプター分子であるmyeloid-differentiation factor 88(MYD88)と結合し,インターロイキン1β受容体のシグナル複合体の一部になっていることはよく知られている57).サイトヘジン・ファミリーは直接的,間接的に受容体直下か細胞膜周辺部に局在し,受容体からのシグナルを可能な限り早く,Arf6に伝える役割があるのかもしれない.興味深いことに,カポジ肉腫の原因であるヘルペスウイルスHHV8にコードされたKaposin Aという2回膜貫通型の推定構造を持つ受容体が宿主細胞のサイトヘジン1に結合し,それがHHV8の病態発現に関与することが明らかにされた58).病態進行過程で,宿主細胞のアクチン細胞骨格系が利用されることも示唆されている59, 60).
BioGRIDウェブサイト上には,50種類以上のサイトヘジン結合候補分子があり,そのほとんどが受容体以外の分子である.しかし,生化学的手法で結合実験が行われた分子は,そのうち10数種類程度である.そのなかで,Cytohesin-associated scaffold protein(CASP,別名CYTIPまたはCybr)とFERM domain containing 4A(FRMD4A)はサイトヘジン1と特異的に結合する.CASPは,そのN末端部分がサイトヘジン1のN末端部分と結合する.この両者の結合配列内にはコイルド・コイルドメインがあり,それがCASPとサイトヘジン1の結合に関与している61–63).FRMD4Aも同様の結合様式をとる64).
コイルド・コイルドメインを介したサイトヘジン・ファミリーと,その結合タンパク質との相互作用は,サイトヘジンの細胞内局在を制御すると考えられている.CASPはSNX27を介して,サイトヘジン1をエンドソームに局在させる65).同様に,RalA binding protein 1(RalBP1)も,コイルド・コイルドメイン間の結合を介し,サイトヘジン2をエンドソームに局在させる66).FRMD4AはPar-3を介して,極性複合体にサイトヘジン1を局在させる64).FRMD4Aと同じファミリー分子であるFRMD4B[別名Grp1 signaling partner 1(GRSP1)]はサイトヘジン3に結合することが知られている67, 68).サイトヘジン3は,細胞極性形成に関与していることが知られている69).また,FRMD4Aは運動失調症の原因遺伝子産物であるため70),FRMD4Bにもその可能性が指摘されている.サイトヘジン2や3の結合タンパク質として同定されたinteraction protein for cytohesin exchange factor 1(IPCEF1)やGrp1-associated scaffold protein(GRASP,別名Tamalin)も,サイトヘジン2および3のN末端部分とIPCEF1のC末端側71)やGRASPのN末端側にあるコイルド・コイルドドメインを介して結合する72, 73).IPCEF1やGRASPは細胞膜の近傍に存在するため,細胞膜直下にサイトヘジン2および3を局在させ,サイトヘジン分子濃度を上昇させる役割があるかもしれない.IPCEF1はサイトヘジン2の活性化に関与することが知られている.
Ras-MAPキナーゼのシグナル経路の足場タンパク質として知られているconnector enhancer of KSR 1および2(CNK1および2)は,サイトヘジン2や3に結合することで,インスリンシグナルの増強に関与している.この結合様式もコイルド・コイルドドメイン間によるものである74, 75).特にCHK1とサイトヘジン3の結合は,インスリンシグナルの増強76, 77)という脂肪細胞における重要な細胞応答に含まれる.
サイトヘジン・ファミリーの基質であるArf6や同じArfファミリー分子のArl4A, Arl4C, Arl4Dもサイトヘジン2と3に結合する78–81).これらの低分子量GTP結合タンパク質がサイトヘジン2や3のC末端にある塩基性アミノ酸に富む配列を含むPHドメイン全体と結合することで,サイトヘジンを細胞膜およびエンドソームにリクルートする.しかし,Arf6やArl4A/C/Dが,サイトヘジン2と3を活性化するかは不明である.
BioGRIDを含む公的なインタラクトーム・ウェブサイトのなかにも,同様の結合様式が推定される分子が多数あり,最近,それが実証されつつある82–85).
4. 受容体下流のミエリン化シグナルの新規メディエーターとしてのサイトヘジン1と,その活性化調節メカニズム
1)ミエリン化開始のシグナルをつかさどるサイトヘジン1
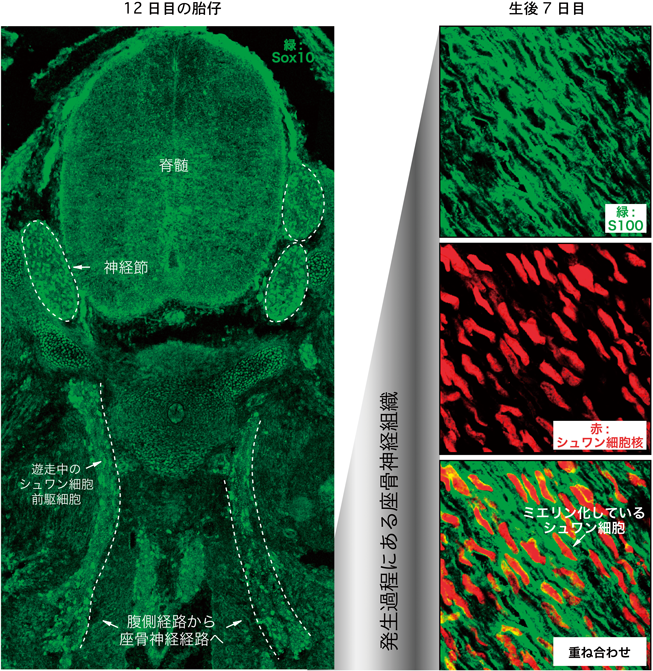
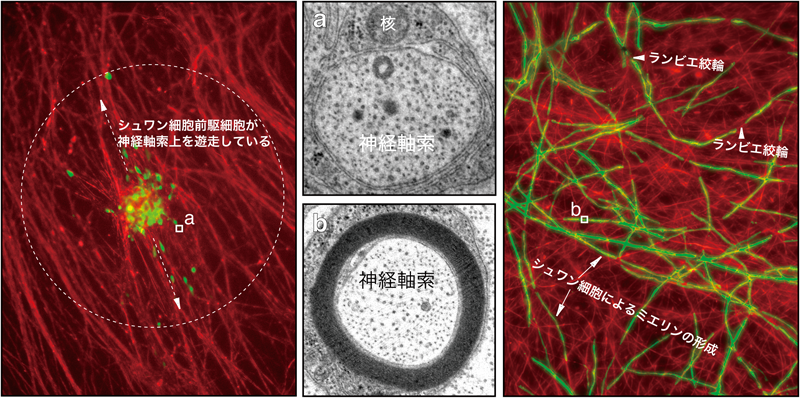
In vitroで有髄神経を作る培養システムが,末梢神経のミエリン化の分子メカニズムの解明に役立ってきた.これは,胎生中期の後根神経節からシュワン細胞前駆細胞と未成熟な神経細胞を別々に単離後精製し,両者を共培養するというものである.通常,この共培養システムは,ミエリン化直前の生後のシュワン細胞を用いるのが普通であるが19, 86, 87),生存能力の強いシュワン細胞前駆細胞を用いることで,その長期培養が可能になった88, 89).これによって,マウスやラットにおける生後2か月までのミエリン発生を,in vivoとほぼ同一のタイムコースで再現できるようになった(図7, 8).また,前ミエリン発生期における前駆細胞の細胞増殖,遊走過程もin vitroで再現させることに成功した.特に,細胞遊走過程は比較的短いタイムコースで観察できる現象なので,顕微鏡システムで極力出力を弱めたレーザーを用いれば,ムービー撮影も可能になる.
この共培養システムを用いて,SecinH3という化合物76, 77)がミエリン化を抑制することが判明した88).SecinH3はミエリン化に影響を及ぼすが,シュワン細胞の数や神経軸索の数および長さを変化させなかった.SecinH3はサイトヘジン・ファミリーに特異的な阻害剤であるが,サイトヘジンの活性中心に結合するため,4種類のサイトヘジンすべてを阻害する.そこで,新生仔由来のシュワン細胞に発現しているサイトヘジンの分子種を調べたところ,サイトヘジン1が豊富に発現していることが判明した.サイトヘジン1に特異的な二本鎖siRNAを作製し,RNA干渉実験を行ったところ,SecinH3のミエリン化に対する抑制効果と同様な結果を得ることができた.これらの研究から,サイトヘジン1は少なくともミエリン発生の初期過程に関与することが示唆された.
そこで,SecinH3のミエリン化への効果がin vivoレベルでも再現できるか明らかにするために,エキソン4から7までを削除したノックアウトマウスを作製した88).当時,サイトヘジン1は血球系や免疫系の細胞のみに発現が確認されていた.また,これらの細胞にはサイトヘジン2も発現していることが知られていた.したがって,古典的ノックアウト法でサイトヘジン1をノックアウトしても胎生致死にならないと判断し,その作製を行った.予想どおり,作製されたマウスは産まれ,外見上の異常も確認されなかった.しかし,座骨神経の輪切り電子顕微鏡写真をとったところ,同腹の野生型マウスの画像と比べると神経軸索のミエリン化率が減少し,ミエリンも薄いことが判明した.他の組織には異常は観察されなかった.また,発生過程の座骨神経の電気伝導速度を測定したところ,脱ミエリン病モデルマウスのような伝導速度の低下はみられなかったものの,有意な伝導速度の低下が検出された.発生過程が進行するにつれて,ノックアウトマウスと野生型マウスの間の伝導速度の差は小さくなった.しかし,完全に差がなくなることはなかった.
これらの現象は,末梢神経ではシュワン細胞特異的なglial fibrillary acidic protein(GFAP)プロモーター下に配置したサイトヘジン1の活性抑制変異体のトランスジェニックマウスの座骨神経でも,同様な結果を得ることができた88).
さらに,これらの遺伝子改変マウスの座骨神経では,サイトヘジン1下流のArf6の活性化が顕著に抑制されることも明らかになった88).遺伝子改変マウスではMPZなどの細胞接着因子の顕著な発現低下は認められたものの,Oct6やKrox20などの転写因子の発現レベルは野生型と変わらなかった.このことは,サイトヘジン1が積極的に細胞の分化過程に関与せず,ミエリン膜の形成に深く関与していることを示唆している.
逆に,野生型のサイトヘジン1のトランスジェニックマウスの座骨神経では,肥厚化したミエリンが観察された90).これは5000倍以上の電子顕微鏡写真から,それぞれのミエリン膜が厚くなったわけではなく,ミエリン膜の巻き数が増加したため,ミエリン全体が肥厚化したように観察された結果であることがわかった.一方,神経軸索の数や直径に変化は観察されなかった.くわえて,Arf6の活性促進,MPZの発現誘導も観察された.トランスジェニックマウスにおいてサイトヘジン1の発現量は2倍であった.
ここでノックアウトマウスおよびトランスジェニックマウスとミエリンの厚さの関係をまとめる(図9).サイトヘジン1は末梢神経組織内ではシュワン細胞に発現しており,その発現量とミエリンの厚さには相関関係がある.ただ,その相関関係には下限と上限があるようだ.サイトヘジン1の発現が完全になくなっても,ミエリンの厚さはゼロにはならない.これは前述した多くのミエリン関連分子がミエリン形成をバックアップしているからであると推定される.一方,サイトヘジン1の発現を増加させても,ミエリン発生期を過ぎてミエリン膜を巻き続けることはなかった.サイトヘジン1はミエリンの厚さの調節因子であるが,その遺伝子改変はマウスの発生に重篤な影響を及ぼさない.もしサイトヘジン1の発現を人為的に制御またはサイトヘジン1に対する化合物でサイトヘジン1の活性調節ができれば,ミエリンの厚さを人為的に制御できるかもしれない.
2)Fynキナーゼによるサイトヘジン1の活性化と,そのミエリン化における役割
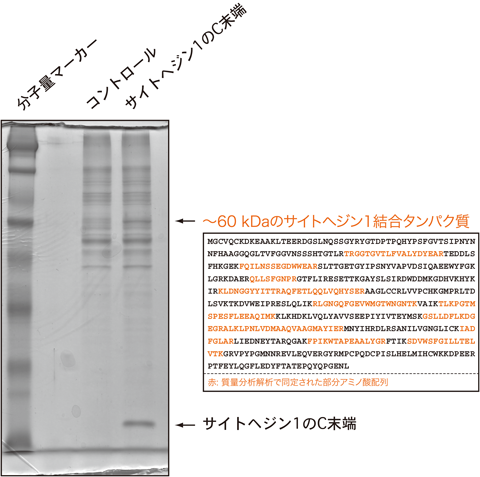
サイトヘジン1のPHドメインとそれに続く塩基性アミノ酸に富む配列をベイトとし,ラットのRT4-D6P2Tシュワン細胞株で,それを発現させ共沈降実験を行った.染色されたゲル上にベイトとの共沈降産物として複数のタンパク質バンドが認められたが,~60 kDaのバンドが特異的にベイトと共沈降することがわかった(図10).質量分析から,このバンドはFynキナーゼであることが判明した88, 91).Fynキナーゼは中枢神経のミエリン化において重要な役割をする分子であることが知られている92).
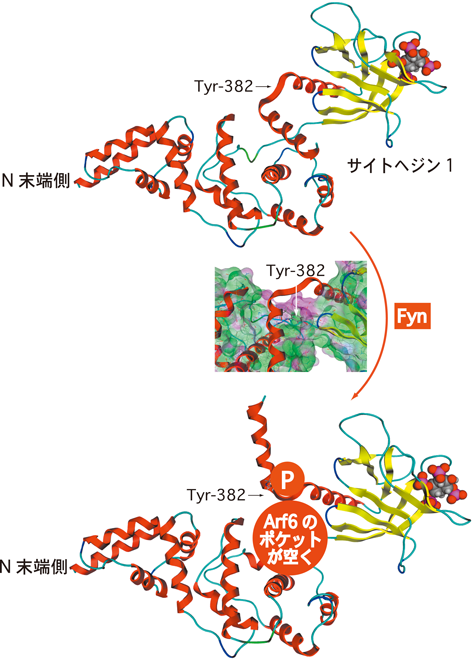
いくつかの生化学実験から,Fynのキナーゼドメインがサイトヘジン1との結合に関与し(Kd値は~88 nM),Fynがサイトヘジン1のC末端側に位置する382番目のチロシン残基(マウスTyr-382)を特異的にリン酸化し(図11),それを活性化することが判明した.これらの現象が初代培養のシュワン細胞においても,その共培養においても,遺伝子改変マウスにおいてもおおむね成立することがわかった.
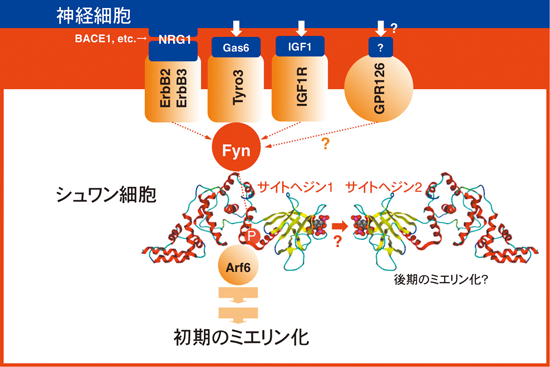
また,Fynは,シュワン細胞のミエリン化の主要な増殖因子受容体であるErbB3/ErbB2受容体とIGF1受容体により活性化されることが明らかになり,その経路上にサイトヘジン1があることが判明した.
当研究室を含んだ国内外のいくつかの研究グループでシュワン細胞のトランスクリプトーム解析がなされた.シュワン細胞の受容体転写産物において,ErbB3/ErbB293)やIGF1受容体と同レベルか,それ以上のレベルでTyro3と呼ばれる増殖因子受容体を持っていることが明らかにされた94).Tyro3の細胞内キナーゼドメインを用いたプロテオーム解析から,そこにFynが結合することも判明した.Tyro3は,ErbB3/ErbB2やIGF1受容体と同様に,Fynを活性化することで,サイトヘジン1の活性化に関与することが示唆された.
サイトヘジン3の結晶構造95)をもとにサイトヘジン1の3D構造を予測することで,Fynによるサイトヘジン1の活性化メカニズムを以下のように推定した.サイトヘジン1のPHドメインに続くC末端の塩基性アミノ酸に富む配列は,サイトヘジン1の触媒部位を覆っている.つまり,定常状態のサイトヘジン1はArf6と接触できない状態にある.コンホメーション変化でC末端部分がこの触媒部位から離れると,サイトヘジン1はArf6を活性化できると推定される.おそらく,このC末端に存在するTyr-382がFynでリン酸化されることにより,C末端とArf6が接触できるようになるのではないかと考えられる.仮想的な活性化型サイトヘジン1の模式図としてサイトヘジン1のC末端の配置を逆転させた図を掲載する(図12).
3)中後期以降のミエリン発生に関与するサイトヘジン2
経時的なトランスクリプトーム解析から,サイトヘジン1の発現量は生後徐々に低下することがわかった.その過程で,サイトヘジン2が1に置き換わるように増加した.このことから,サイトヘジン2が中後期のミエリン発生を制御すると考えられた.そこで,サイトヘジン2のコンディショナル・ノックアウトマウスを作製した96).サイトヘジン2は普遍的に発現しているため,エキソン6と7をloxP配列ではさんだかたちでマウスを作製し,シュワン細胞特異的Creマウスと交配後,座骨神経のミエリンを調べた.その結果,コントロールマウスの座骨神経に比べて,ノックアウトマウスではミエリン化率もミエリンの厚さも薄いことが判明した.しかし,神経軸索へのノックアウトの影響は観察されなかった.今後,サイトヘジン2が中後期以降のArf6の活性を担う分子であるのか,また,それがサイトヘジン1と類似したシグナル伝達経路上にあるか検討が必要である(図13).
サイトヘジン1の上流のFynおよび下流のArf6の機能欠失型遺伝子改変マウスでは,サイトヘジン2と類似した形質を示すことがわかっている91, 97, 98).
5. シュワン細胞前駆細胞におけるサイトヘジン1の発現とその役割
サイトヘジン1は胎生期の未成熟な末梢神経節や座骨神経にも発現している.胎生期では,シュワン細胞前駆細胞が神経軸索上で細胞分裂を繰り返しながら,巻くべき軸索の位置を探すために軸索上を遊走し,ミエリン化の準備をする.マウスのシュワン細胞は胎生12日目まで前駆細胞である.12日を超えると細胞分裂速度も細胞遊走能も徐々に低下し,未成熟シュワン細胞になる.サイトヘジン1のノックアウトマウスでは,胎生12日目においても前駆細胞の遊走遅延が観察された99).また,細胞遊走過程でも「Fynキナーゼ→サイトヘジン1→Arf6」経路が重要な役割を持つことが判明した.一方,未成熟シュワン細胞がシュワン細胞へと成熟するにつれ,ノックアウトマウスでみられた遊走遅延は観察されなくなった.
6. オリゴデンドロサイトのミエリン発生におけるサイトヘジン2のユニークな分子メカニズム
オリゴデンドロサイトがミエリンを作る過程においても,サイトヘジンが関与していることが明らかになっている.オリゴデンドロサイトにはサイトヘジン2が豊富に発現している.しかし,オリゴデンドロサイトのサイトヘジン2のシグナルネットワークはシュワン細胞と異なっており,きわめて特徴的である100).オリゴデンドロサイトでは,受容体シグナルによって「サイトヘジン2→Arf6」経路が積極的に活性化されミエリン化が促進されるのではなく,この経路の抑制シグナルが解除され,その結果としてArf6が活性化されミエリン化が促進する.この抑制シグナルにはRab35とArfGAP with coiled-coil, ankyrin repeat and PH domains 2(ACAP2)が関与している.Rab35はRabファミリーの低分子量GTP結合タンパク質であり,その下流分子の一つがACAP2である.ACAP2はArf6を負に制御する分子である.Rab35とACAP2の活性はオリゴデンドロサイトのミエリン化とともに減弱するため,ACAP2の活性が低下すれば,相対的に,Arf6の不活性化メカニズムが解除され「サイトヘジン2→Arf6」経路が強調される.ただし,何が引き金になりRab35とACAP2の活性が減弱するのか不明である.前述した「オリゴデンドロサイト・タイマー」が,それを制御しているのかもしれない.くわえて,神経細胞のArf6も間接的にミエリン化を制御しすることが知られており,Arf6はオリゴデンドロサイト内外でミエリン化を促進する役割を持っている101).
今日ではCharcot-Marie-Tooth病(シャルコー・マリー・トゥース病,CMT病)は先天性の末梢神経変性症の総称として用いられることが多くなった.CMT病の有病率は2500人に1人程度であり,その原因遺伝子の数は50種類を超えている.CMT病の原因遺伝子の異常は,点変異,重複,欠損など,さまざまなタイプの変異が確認されているが,最も多い変異は点変異である.点変異による遺伝子産物の機能欠失が原因で,CMT病の病態が発現する場合が多い.
CMT病には,いくつかの病型がある.1型は全有病率の50%以上を占める.1型の遺伝子変異(優性変異)はシュワン細胞に異常を起こす.そのため,脱ミエリンと再ミエリン化を繰り返し,最終的に,ほとんどの神経線維が脱ミエリン状態になる.神経伝導速度が遅延(神経伝導速度が38 m/secより遅い)し,歩行困難に陥るケースもある.
2型(優性変異)は全有病率の20~40%を占める.2型の遺伝子変異は末梢神経細胞に異常を起こす.神経伝導速度の遅延は認められない(神経伝導速度が38 m/secより速い)か,ごくわずかの低下がみられる程度である.病態が進行すれば歩行困難になる.
4型は劣性変異の疾患として分類されているが,その遺伝子変異はシュワン細胞に異常を起こす場合が多い.病態の進行は比較的遅い.その他のCMT病は,X染色体に原因遺伝子があるCMTX(6型まで同定されている)やCMT, dominant intermediate(CMTDI)(F型まで同定されている)などがある.国内外の複数の研究グループが,次世代シークエンサーを用い,末梢神経変性病態を示す疾患の遺伝子解析を行っている.今後,多くのCMT病関連の原因遺伝子が発見されるだろう.
最初に見つかったCMT病の原因遺伝子cmt1aはシュワン細胞に豊富に発現しているPMP22をコードしている.PMP22の変異で起きるCMT1A病は,CMT病のなかで最も多い原因である.CMT1A病は,他のCMT病と同様に,特異的な治療薬がない.一方,基礎研究は比較的進んでおり,優れた疾患モデル動物(マウスやラットなど)が作製されている9–12).そこで,ミエリン発生の促進分子であるサイトヘジン1がCMT1A病の脱ミエリン現象を改善できるか検討した.サイトヘジン1のトランスジェニックマウスとCMTIA型の疾患モデルマウスを交配し,産仔の座骨神経を解析したところ,疾患モデルマウスのミエリンの厚さが野生型に近いレベルまで戻った.しかし,すべての産仔でこれが確認されたわけではない.くわえて,滑面上でのマウスの歩行も改善傾向にあった.今後,詳細な運動機能解析および行動試験を行う必要がある.
以上の結果は,サイトヘジン1がCMT1A病の創薬標的分子であることを期待させる.しかし,この場合,サイトヘジン1の過剰発現により病態改善が観察された.一般的に酵素活性の促進薬を作ることは難しい.サイトヘジン1はその活性化メカニズムが明らかであるため,促進性の化合物を開発できる可能性はある.くわえて,サイトヘジン1の発現パターンは全身性ではなく,きわめて特異的である.そのため標的分子として適切である.これは,オリゴデンドロサイトに起因する疾患改善研究で,全身発現性のサイトヘジン2を利用することが容易ではないことと対照的である.また,サイトヘジン1の考えられる限りの遺伝子改変マウスを作製しても,外見上の異常はみられない.サイトヘジン1はシュワン細胞に起因する末梢神経変性疾患の治療標的候補の一つであると期待される.
以上の研究は,遺伝子改変マウスを用い,どのようにミエリンの「厚さ」が形成されるかに絞ったものであった.しかし,神経線維は2Dでなく3Dである.ミエリン構造を正確に検討するためには,神経線維の連続切片の画像取得とデータ再構築による3D構造解析が必要になるだろう.
くわえて,ミエリン維持やミエリン再構築の共通の分子メカニズムを解明することは今後の研究課題の一つである.特に,オリゴデンドロサイトのミエリン化は柔軟性に富むようだ.神経細胞の活動電位の変化や神経伝達物質の放出サイクルとミエリン化は密接な関係がある102).さらに,神経領域ごとに異なったオリゴデンドロサイトがあるという報告があり,それが神経領域ごとのミエリン化の状態や維持と関係があるのか,他の機能と関係しているのかは重要な問題である103).
一方,先天性の脱ミエリン疾患における創薬標的分子の探索研究104–109)が,炎症や環境要因など複雑な発症原因をもつ脱ミエリン疾患の研究に応用可能かどうか考える必要がある110, 111).脱ミエリン疾患に共通の創薬標的分子があるかどうか今後の研究が待たれる.
いずれの研究においても,マウス以上のモデル動物を用いることで初めて,ミエリンの構造,発生,維持,再編成,疾患の研究がヒトと比較可能な状態で達成されると期待される.また,そのためには,ヒト型の有髄組織の研究を遂行させる実験システムの構築も重要かもしれない.
本稿では,ヒト遺伝子やマウス遺伝子,および,その産物に関して,それぞれ大文字や小文字などの統一した表記を用いず,最も一般的に使用されている方法で分子名を記載した.また,ミエリン発生の分子メカニズムに関する研究は発展途上であるため,限定的な表現を使用した箇所も多く,ご容赦願いたい.
謝辞Acknowledgments
2009年に本誌に総説を書く機会をいただきました.その後,ミエリン発生の研究は飛躍的に進歩しました.最近の国内外の研究内容すべてを本稿に記載することができませんでした.これに関してもご容赦頂ければ幸いです.
さいごに,本研究を遂行するにあたり,国立研究開発法人国立成育医療研究センター研究所の松原洋一所長,斎藤博久副所長,梅澤明弘副所長,田上昭人部長のひとかたならぬご高配に感謝いたします.本研究は,研究所薬剤治療研究部の宮本幸さん,ベイラー大学医学部の鳥居知宏さんを始めとした多くの皆様によって支えられており,この場を借りて感謝の言葉に代えさせていただきます.
引用文献References
1) Bunge, R.P., Bunge, M.B., & Eldridge, C.F. (1986) Annu. Rev. Neurosci., 9, 305–328.
2) Bunge, R.P. (1993) Curr. Opin. Neurobiol., 3, 805–809.
3) Jessen, K.R. & Mirsky, R. (2005) Nat. Rev. Neurosci., 6, 671–682.
4) Jessen, K.R., Mirsky, R., & Lloyd, A.C. (2015) Cold Spring Harb. Perspect. Biol., 7, a020487.
5) Salzer, J.L., Brophy, P.J., & Peles, E. (2008) Glia, 56, 1532–1534.
6) Salzer, J.L. (2015) Cold Spring Harb. Perspect. Biol., 7, a020529.
7) Nave, K.A. & Salzer, J.L. (2006) Curr. Opin. Neurobiol., 16, 492–500.
8) Nave, K.A. & Werner, H.B. (2014) Annu. Rev. Cell Dev. Biol., 30, 503–533.
9) Taveggia, C., Feltri, M.L., & Wrabetz, L. (2010) Nat. Rev. Neurol., 6, 276–287.
10) Monk, K.R., Feltri, M.L., & Taveggia, C. (2015) Glia, 63, 1376–1393.
11) Sango, K. & Yamauchi, J.(2014) Schwann Cell Development and Pathology (Sango, K., Yamauchi, J., Eds.), pp. 1–4, Springer.
12) Miyamoto, Y. & Yamauchi, J.(2014) Schwann Cell Development and Pathology (Sango, K., Yamauchi, J., Eds.), pp. 5–27, Springer.
13) Tomassy, G.S., Berger, D.R., Chen, H.H., Kasthuri, N., Hayworth, K.J., Vercelli, A., Seung, H.S., Lichtman, J.W., & Arlotta, P. (2014) Science, 344, 319–324.
14) Freeman, S.A., Desmazières, A., Simonnet, J., Gatta, M., Pfeiffer, F., Aigrot, M.S., Rappeneau, Q., Guerreiro, S., Michel, P.P., Yanagawa, Y., Barbin, G., Brophy, P.J., Fricker, D., Lubetzki, C., & Sol-Foulon, N. (2015) Proc. Natl. Acad. Sci. USA, 112, E321–E328.
15) Masaki, T., Qu, J., Cholewa-Waclaw, J., Burr, K., Raaum, R., & Rambukkanam, A. (2013) Cell, 152, 51–67.
16) Fünfschilling, U., Supplie, L.M., Mahad, D., Boretius, S., Saab, A.S., Edgar, J., Brinkmann, B.G., Kassmann, C.M., Tzvetanova, I.D., Möbius, W., Diaz, F., Meijer, D., Suter, U., Hamprecht, B., Sereda, M.W., Moraes, C.T., Frahm, J., Goebbels, S., & Nave, K.A. (2012) Nature, 485, 517–521.
17) Dyachuk, V., Furlan, A., Shahidi, M.K., Giovenco, M., Kaukua, N., Konstantinidou, C., Pachnis, V., Memic, F., Marklund, U., Müller, T., Birchmeier, C., Fried, K., Ernfors, P., & Adameyko, I. (2014) Science, 345, 82–87.
18) Espinosa-Medina, I., Outin, E., Picard, C.A., Chettouh, Z., Dymecki, S., Consalez, G.G., Coppola, E., & Brunet, J.F. (2014) Science, 345, 87–90.
19) Chan, J.R., Jolicoeur, C., Yamauchi, J., Elliott, J., Fawcett, J.P., Ng, B.K., & Cayouette, M. (2006) Science, 314, 832–836.
20) Chan, J.R. (2007) J. Cell Biol., 177, 953–955.
21) Wegner, M. & Stolt, C.C. (2005) Trends Neurosci., 28, 583–588.
22) Stolt, C.C. & Wegner, M. (2016) Brain Res., 1641(Pt A), 101–110.
23) Britsch, S., Goerich, D.E., Riethmacher, D., Peirano, R.I., Rossner, M., Nave, K.A., Birchmeier, C., & Wegner, M. (2001) Genes Dev., 15, 66–78.
24) Finzsch, M., Schreiner, S., Kichko, T., Reeh, P., Tamm, E.R., Bösl, M.R., Meijer, D., & Wegner, M. (2010) J. Cell Biol., 189, 701–712.
25) He, D., Marie, C., Zhao, C., Kim, B., Wang, J., Deng, Y., Clavairoly, A., Frah, M., Wang, H., He, X., Hmidan, H., Jones, B.V., Witte, D., Zalc, B., Zhou, X., Choo, D.I., Martin, D.M., Parras, C., & Lu, Q.R. (2016) Nat. Neurosci., 19, 678–689.
26) Emery, B., Agalliu, D., Cahoy, J.D., Watkins, T.A., Dugas, J.C., Mulinyawe, S.B., Ibrahim, A., Ligon, K.L., Rowitch, D.H., & Barres, B.A. (2009) Cell, 138, 172–185.
27) Schwab, M.E. (2004) Curr. Opin. Neurobiol., 14, 118–240.
28) von Büdingen, H.C., Mei, F., Greenfield, A., Jahn, S., Shen, Y.A., Reid, H.H., McKemy, D.D., & Chan, J.R. (2015) J. Cell Biol., 210, 891–898.
29) Penzes, P., Buonanno, A., Passafaro, M., Sala, C., & Sweet, R.A. (2013) J. Neurochem., 126, 165–182.
30) Dugas, J.C., Ibrahim, A., & Barres, B.A. (2007) J. Neurosci., 27, 6185–6196.
31) Ogata, T., Iijima, S., Hoshikawa, S., Miura, T., Yamamoto, S., Oda, H., Nakamura, K., & Tanaka, S. (2004) J. Neurosci., 24, 6724–6732.
32) Yamauchi, J., Miyamoto, Y., Chan, J.R., & Tanoue, A. (2008) J. Cell Biol., 181, 351–365.
33) Miyamoto, Y., Yamauchi, J., Chan, J.R., Okada, A., Tomooka, Y., Hisanaga, S., & Tanoue, A. (2007) J. Cell Sci., 120, 4355–4366.
34) Miyamoto, Y., Yamauchi, J., & Tanoue, A. (2008) J. Neurosci., 28, 8326–8337.
35) Cheng, C.M., Joncas, G., Reinhardt, R.R., Farrer, R., Quarles, R., Janssen, J., McDonald, M.P., Crawley, J.N., Powell-Braxton, L., & Bondy, C.A. (1998) J. Neurosci., 18, 5673–5681.
36) Sweeney, M.D., Ayyadurai, S., & Zlokovic, B.V. (2016) Nat. Neurosci., 19, 771–783.
37) Monk, K.R., Naylor, S.G., Glenn, T.D., Mercurio, S., Perlin, J.R., Dominguez, C., Moens, C.B., & Talbot, W.S. (2009) Science, 325, 1402–1405.
38) Paavola, K.J., Sidik, H., Zuchero, J.B., Eckart, M., & Talbot, W.S. (2014) Sci. Signal., 7, ra76.
39) Petersen, S.C., Luo, R., Liebscher, I., Giera, S., Jeong, S.J., Mogha, A., Ghidinelli, M., Feltri, M.L., Schöneberg, T., Piao, X., & Monk, K.R. (2015) Neuron, 85, 755–769.
40) Lyons, D.A. & Talbot, W.S. (2014) Cold Spring Harb. Perspect. Biol., 7, a020586.
41) Dixon, B., Mansour, M., & Pohajdak, B. (1993) Cytogenet. Cell Genet., 63, 42–44.
42) Kolanus, W., Nagel, W., Schiller, B., Zeitlmann, L., Godar, S., Stockinger, H., & Seed, B. (1996) Cell, 86, 233–242.
43) Meacci, E., Tsai, S.C., Adamik, R., Moss, J., & Vaughan, M. (1997) Proc. Natl. Acad. Sci. USA, 94, 1745–1758.
44) Goda, N., Tanoue, A., Kikuchi, S., & Tsujimoto, G. (2000) Biochim. Biophys. Acta, 1493, 195–199.
45) Cherfils, J., Ménétrey, J., Mathieu, M., Le Bras, G., Robineau, S., Béraud-Dufour, S., Antonny, B., & Chardin, P. (1998) Nature, 392, 101–105.
46) Frank, S., Upender, S., Hansen, S.H., & Casanova, J.E. (1998) J. Biol. Chem., 273, 23–27.
47) Klarlund, J.K., Guilherme, A., Holik, J.J., Virbasius, J.V., Chawla, A., & Czech, M.P. (1997) Science, 275, 1927–1930.
48) Franco, M., Boretto, J., Robineau, S., Monier, S., Goud, B., Chardin, P., & Chavrier, P. (1998) Proc. Natl. Acad. Sci. USA, 95, 9926–9931.
49) Ogasawara, M., Kim, S.C., Adamik, R., Togawa, A., Ferrans, V.J., Takeda, K., Kirby, M., Moss, J., & Vaughan, M. (2000) J. Biol. Chem., 275, 3221–3230.
50) Klarlund, J.K., Tsiaras, W., Holik, J.J., Chawla, A., & Czech, M.P. (2000) J. Biol. Chem., 275, 32816–32821.
51) Cronin, T.C., DiNitto, J.P., Czech, M.P., & Lambright, D.G. (2004) EMBO J., 23, 3711–3720.
52) Weber, K.S., Weber, C., Ostermann, G., Dierks, H., Nagel, W., & Kolanus, W. (2001) Curr. Biol., 11, 1969–1974.
53) Casanova, J.E. (2007) Traffic, 8, 1476–1485.
54) Mizuno-Yamasaki, E., Rivera-Molina, F., & Novick, P. (2012) Annu. Rev. Biochem., 81, 637–659.
55) Quast, T., Tappertzhofen, B., Schild, C., Grell, J., Czeloth, N., Förster, R., Alon, R., Fraemohs, L., Dreck, K., Weber, C., Lämmermann, T., Sixt, M., & Kolanus, W. (2009) Blood, 113, 5801–5810.
56) Oh, S.J. & Santy, L.C. (2010) J. Biol. Chem., 285, 14610–14616.
57) Zhu, W., London, N.R., Gibson, C.C., Davis, C.T., Tong, Z., Sorensen, L.K., Shi, D.S., Guo, J., Smith, M.C., Grossmann, A.H., Thomas, K.R., & Li, D.Y. (2012) Nature, 492, 252–255.
58) Kliche, S., Nagel, W., Kremmer, E., Atzler, C., Ege, A., Knorr, T., Koszinowski, U., Kolanus, W., & Haas, J. (2001) Mol. Cell, 7, 833–834.
59) Dierks, H., Kolanus, J., & Kolanus, W. (2001) J. Biol. Chem., 276, 37472–37481.
60) Humphreys, D., Davidson, A.C., Hume, P.J., Makin, L.E., & Koronakis, V. (2013) Proc. Natl. Acad. Sci. USA, 110, 16880–16885.
61) Mansour, M., Lee, S.Y., & Pohajdak, B. (2002) J. Biol. Chem., 277, 32302–32309.
62) Boehm, T., Hofer, S., Winklehner, P., Kellersch, B., Geiger, C., Trockenbacher, A., Neyer, S., Fiegl, H., Ebner, S., Ivarsson, L., Schneider, R., Kremmer, E., Heufler, C., & Kolanus, W. (2003) EMBO J., 22, 1014–1024.
63) Watford, W.T., Li, D., Agnello, D., Durant, L., Yamaoka, K., Yao, Z.J., Ahn, H.J., Cheng, T.P., Hofmann, S.R., Cogliati, T., Chen, A., Hissong, B.D., Husa, M.R., Schwartzberg, P., O’Shea, J.J., & Gadina, M. (2006) Mol. Cell. Biol., 26, 6623–6632.
64) Ikenouchi, J. & Umeda, M. (2010) Proc. Natl. Acad. Sci. USA, 107, 748–753.
65) MacNeil, A.J., Mansour, M., & Pohajdak, B. (2007) Biochem. Biophys. Res. Commun., 359, 848–853.
66) Wurtzel, J.G., Lee, S., Singhal, S.S., Awasthi, S., Ginsberg, M.H., & Goldfinger, L.E. (2015) Biochem. Biophys. Res. Commun., 467, 785–791.
67) Klarlund, J.K., Holik, J., Chawla, A., Park, J.G., Buxton, J., & Czech, M.P. (2001) J. Biol. Chem., 276, 40065–40070.
68) DiNitto, J.P., Lee, M.T., Malaby, A.W., & Lambright, D.G. (2010) Biochemistry, 49, 6083–6092.
69) Teuliere, J., Cordes, S., Singhvi, A., Talavera, K., & Garriga, G. (2014) Genetics, 198, 229–247.
70) Fine, D., Flusser, H., Markus, B., Shorer, Z., Gradstein, L., Khateeb, S., Langer, Y., Narkis, G., Birk, R., Galil, A., Shelef, I., & Birk, O.S. (2015) Eur. J. Hum. Genet., 23, 1729–1734.
71) Venkateswarlu, K. (2003) J. Biol. Chem., 278, 43460–43469.
72) Nevrivy, D.J., Peterson, V.J., Avram, D., Ishmael, J.E., Hansen, S.G., Dowell, P., Hruby, D.E., Dawson, M.I., & Leid, M. (2000) J. Biol. Chem., 275, 16827–16836.
73) Kitano, J., Kimura, K., Yamazaki, Y., Soda, T., Shigemoto, R., Nakajima, Y., & Nakanishi, S. (2002) J. Neurosci., 22, 1280–1289.
74) Lim, J., Zhou, M., Veenstra, T.D., & Morrison, D.K. (2010) Genes Dev., 24, 1496–1506.
75) Lim, J., Ritt, D.A., Zhou, M., & Morrison, D.K. (2014) Curr. Biol., 24, 786–792.
76) Hafner, M., Schmitz, A., Grüne, I., Srivatsan, S.G., Paul, B., Kolanus, W., Quast, T., Kremmer, E., Bauer, I., & Famulok, M. (2006) Nature, 444, 941–944.
77) Fuss, B., Becker, T., Zinke, I., & Hoch, M. (2006) Nature, 444, 945–948.
78) Cohen, L.A., Honda, A., Varnai, P., Brown, F.D., Balla, T., & Donaldson, J.G. (2007) Mol. Biol. Cell, 18, 2244–2253.
79) Hofmann, I., Thompson, A., Sanderson, C.M., & Munro, S. (2007) Curr. Biol., 17, 711–716.
80) Li, C.C., Chiang, T.C., Wu, T.S., Pacheco-Rodriguez, G., Moss, J., & Lee, F.J. (2007) Mol. Biol. Cell, 18, 4420–4437.
81) Yamauchi, J., Miyamoto, Y., Torii, T., Mizutani, R., Nakamura, K., Sanbe, A., Koide, H., Kusakawa, S., & Tanoue, A. (2009) Exp. Cell Res., 315, 2043–2052.
82) Torii, T., Miyamoto, Y., Sanbe, A., Nishimura, K., Yamauchi, J., & Tanoue, A. (2010) J. Biol. Chem., 285, 24270–24281.
83) Torii, T., Miyamoto, Y., Nishimura, K., Nakamura, K., Maeda, M., Tanoue, A., & Yamauchi, J. (2012) Adv. Biol. Chem., 2, 291–300.
84) Torii, T., Miyamoto, Y., Nakamura, K., Maeda, M., Yamauchi, J., & Tanoue, A. (2012) Cell. Signal., 24, 1872–1882.
85) Torii, T., Miyamoto, Y., Tago, K., Sango, K., Nakamura, K., Sanbe, A., Tanoue, A., & Yamauchi, J. (2014) J. Biol. Chem., 289, 33887–33903.
86) Yamauchi, J., Miyamoto, Y., Hamasaki, H., Sanbe, A., Kusakawa, S., Nakamura, A., Tsumura, H., Maeda, M., Nemoto, N., Kawahara, K., Torii, T., & Tanoue, A. (2011) J. Neurosci., 31, 12579–12592.
87) Torii, T., Miyamoto, Y., Nagao, M., Onami, N., Tsumura, H., Maeda, M., Nakamura, K., Tanoue, A., & Yamauchi, J. (2012) Am. J. Mol. Biol., 2, 210–216.
88) Yamauchi, J., Miyamoto, Y., Torii, T., Takashima, S., Kondo, K., Kawahara, K., Nemoto, N., Chan, J.R., Tsujimoto, G., & Tanoue, A. (2012) Sci. Signal., 5, ra69.
89) Miyamoto, Y., Torii, T., Yamamori, N., Ogata, T., Tanoue, A., & Yamauchi, J. (2013) Sci. Signal., 6, ra15.
90) Torii, T., Miyamoto, Y., Onami, N., Tsumura, H., Nemoto, N., Kawahara, K., Kato, M., Kotera, J., Nakamura, K., Tanoue, A., & Yamauchi, J. (2013) J. Mol. Neurosci., 51, 522–531.
91) Miyamoto, Y., Tamano, M., Torii, T., Kawahara, K., Nakamura, K., Tanoue, A., Takada, S., & Yamauchi, J. (2016) Data Brief, 7, 1098–1105.
92) Umemori, H., Sato, S., Yagi, T., Aizawa, S., & Yamamoto, T. (1994) Nature, 367, 572–576.
93) Torii, T., Miyamoto, Y., Takada, S., Tsumura, H., Arai, M., Nakamura, K., Ohbuchi, K., Yamamoto, M., Tanoue, A., & Yamauchi, J. (2014) Biochem. Biophys. Res. Commun., 452, 782–788.
94) Miyamoto, Y., Torii, T., Takada, S., Ohno, N., Saitoh, Y., Nakamura, K., Ito, A., Ogata, T., Terada, N., Tanoue, A., & Yamauchi, J. (2015) Mol. Biol. Cell, 26, 3489–3503.
95) DiNitto, J.P., Delprato, A., Gabe Lee, M.T., Cronin, T.C., Huang, S., Guilherme, A., Czech, M.P., & Lambright, D.G. (2007) Mol. Cell, 28, 569–583.
96) Torii, T., Ohno, N., Miyamoto, Y., Kawahara, K., Saitoh, Y., Nakamura, K., Takashima, S., Sakagami, H., Tanoue, A., & Yamauchi, J. (2015) Biochem. Biophys. Res. Commun., 460, 819–825.
97) Torii, T., Miyamoto, Y., Yamamoto, M., Ohbuchi, K., Tsumura, H., Kawahara, K., Tanoue, A., Sakagami, H., & Yamauchi, J. (2015) Biochem. Biophys. Res. Commun., 465, 450–457.
98) Torii, T., Miyamoto, Y., Yamamoto, M., Ohbuchi, K., Tsumura, H., Kawahara, K., Tanoue, A., Sakagami, H., & Yamauchi, J. (2015) Data Brief, 5, 388–395.
99) Miyamoto, Y., Torii, T., Nakamura, K., Takashima, S., Sanbe, A., Tanoue, A., & Yamauchi, J. (2013) Cell. Signal., 25, 1379–1387.
100) Miyamoto, Y., Yamamori, N., Torii, T., Tanoue, A., & Yamauchi, J. (2014) Mol. Biol. Cell, 25, 1532–1542.
101) Akiyama, M., Hasegawa, H., Hongu, T., Frohman, M.A., Harada, A., Sakagami, H., & Kanaho, Y. (2014) Nat. Commun., 5, 4744.
102) Koudelka, S., Voas, M.G., Almeida, R.G., Baraban, M., Soetaert, J., Meyer, M.P., Talbot, W.S., & Lyons, D.A. (2016) Curr. Biol., 26, 1447–1455.
103) Marques, S., Zeisel, A., Codeluppi, S., van Bruggen, D., Mendanha Falcão, A., Xiao, L., Li, H., Häring, M., Hochgerner, H., Romanov, R.A., Gyllborg, D., Muñoz-Manchado, A.B., La Manno, G., Lönnerberg, P., Floriddia, E.M., Rezayee, F., Ernfors, P., Arenas, E., Hjerling-Leffler, J., Harkany, T., Richardson, W.D., Linnarsson, S., & Castelo-Branco, G. (2016) Science, 352, 1326–1329.
104) Miyamoto, Y., Torii, T., Tanoue, A., & Yamauchi, J. (2012) Biochem. Biophys. Res. Commun., 424, 262–268.
105) Torii, T., Miyamoto, Y., Yamauchi, J., & Tanoue, A. (2014) Pediatr. Int., 56, 659–666.
106) Miyamoto, Y., Torii, T., Eguchi, T., Nakamura, K., Tanoue, A., & Yamauchi, J. (2014) J. Clin. Neurosci., 21, 1033–1039.
107) Miyamoto, Y., Eguchi, T., Kawahara, K., Hasegawa, N., Nakamura, K., Funakoshi-Tago, M., Tanoue, A., Tamura, H., & Yamauchi, J. (2015) Biochem. Biophys. Res. Commun., 462, 275–281.
108) Miyamoto, Y., Funakoshi-Tago, M., Hasegawa, N., Eguchi, T., Tanoue, A., Tamura, H., & Yamauchi, J. (2016) Data Brief, 6, 482–488.
109) Torii, T. & Yamauchi, J. (2016) Neural Regen. Res., 11, 215–216.
110) Gibson, E.M., Purger, D., Mount, C.W., Goldstein, A.K., Lin, G.L., Wood, L.S., Inema, I., Miller, S.E., Bieri, G., Zuchero, J.B., Barres, B.A., Woo, P.J., Vogel, H., & Monje, M. (2014) Science, 344, 1252304.
111) McKenzie, I.A., Ohayon, D., Li, H., de Faria, J.P., Emery, B., Tohyama, K., & Richardson, W.D. (2014) Science, 346, 318–322.
著者紹介Author Profile
 山内 淳司(やまうち じゅんじ)
山内 淳司(やまうち じゅんじ)東京薬科大学生命科学部分子神経科学研究室教授.理学博士.
略歴1997年東京工業大学大学院生命理工学研究科卒業.同年同ポスドク.2000年国立小児病院ポスドク.01年奈良先端科学技術大学院大学助手.03年スタンフォード大学Visiting Assistant Professor.05年より国立成育医療研究センター研究所室長.08年東京工業大学大学院連携准教授を併任.12年東京医科歯科大学大学院連携教授を併任.16年現職.
研究テーマと抱負末梢神経と中枢神経のミエリン発生の分子メカニズムの研究および先天性のミエリン形成不全疾患の創薬標的分子の探索研究.ミエリン研究や稀少疾患に関する研究を,一般性の高い神経科学研究に応用できるような一本の道筋をつくることに貢献したい.
趣味ムービーファイルを集めること,散歩.