1)細胞競合の発見とその定義
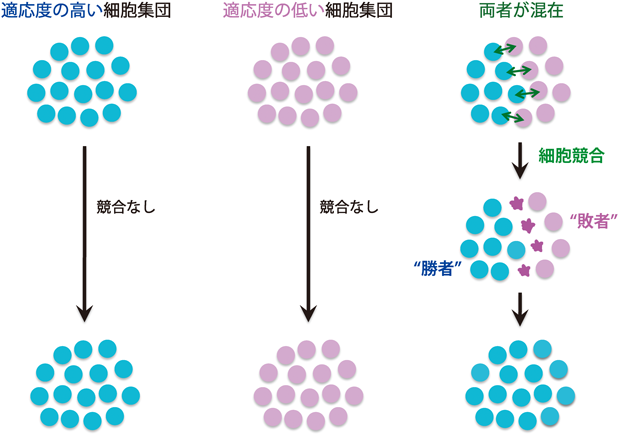
細胞競合現象は,1975年にMorataとRipollによるショウジョウバエMinute変異体の解析を通じて最初に報告された1).Minute変異体とは,リボソームタンパク質遺伝子の機能欠失変異をヘテロに持つ一連のショウジョウバエ変異体の総称で,いずれも発生速度の遅延と剛毛が細くなるという表現型以外には一見正常な個体である2).Morataらは,遺伝的モザイク法を用いてショウジョウバエ翅成虫原基の組織中にMinuteへテロ変異細胞と野生型細胞が混在する状況を作り出した.その結果,発生過程でMinuteへテロ変異細胞が組織から排除され,最終的に野生型細胞のみからなる翅が形成されることを見いだした.Minuteへテロ変異細胞は単独では正常な翅を形成できることから,この変異細胞は野生型細胞との生存競争,すなわちcell competition(細胞競合)の敗者となって組織から排除されると解釈された1).さらにMorataとSimpsonは,翅成虫原基でMinuteへテロ変異細胞と野生型細胞それぞれのクローン成長過程を詳細に解析し,野生型細胞クローンに接していないMinuteへテロ変異細胞は排除されないこと,またMinuteへテロ変異細胞クローンに接した野生型細胞は接していない野生型細胞よりも増殖率が高いことを報告した3).これらの解析から,「組織中で近接する細胞間で相対的に環境適応度の高い細胞が低い細胞を集団から排除する」という,細胞競合のコンセプトが提示された(図1).
Morataらの古典的解析から約30年を経た2002年,Morata, Moreno, Baslerらは部位特異的な体細胞組換え技術による改良型遺伝的モザイク法4)を用い,野生型細胞に接したMinuteへテロ変異細胞が実際に細胞死(アポトーシス)を起こして組織から排除されることを示した5).さらに,この細胞死誘導のメカニズムとして,Minuteへテロ変異細胞は周辺の野生型細胞よりも分泌性生存因子Dppを捕獲する能力が低く,それが起因となって細胞内ストレスキナーゼJNKが活性化して細胞死が起こるという「リガンド捕獲モデル」を提唱した5).しかし,このDppに依存した細胞死誘導のモデルは後にBakerらのMinute細胞競合の遺伝学的解析によって疑義が提出されている6).一方Bakerらは,Minuteへテロ変異細胞の細胞死は近接する野生型細胞による貪食によって引き起こされることを示したが7),Morenoらは貪食は敗者の細胞死の後に主にヘモサイトによって引き起こされるとしてこれに異を唱えている8).キメラマウスを用いた解析により,リボソームタンパク質遺伝子のヘテロ変異(Bst変異)細胞が発生過程で排除されることが報告され9),Minute変異による細胞競合が種を超えて保存された現象であることが示されている.
細胞競合の定義については,いまだ明確なものは存在しない.Bakerによって「細胞非自律的な細胞死誘導」というシンプルかつ合理的な定義づけの提案がなされたものの10),最近では細胞競合の敗者が細胞死を起こすことなく管腔側に押し出されるという哺乳類細胞での知見も蓄積してきており(後述),今後は多様な細胞競合現象とそのメカニズムに基づいた新たな定義づけが必要になってくるかもしれない.(「追記」参照)
2)がん原性細胞による正常細胞の駆逐
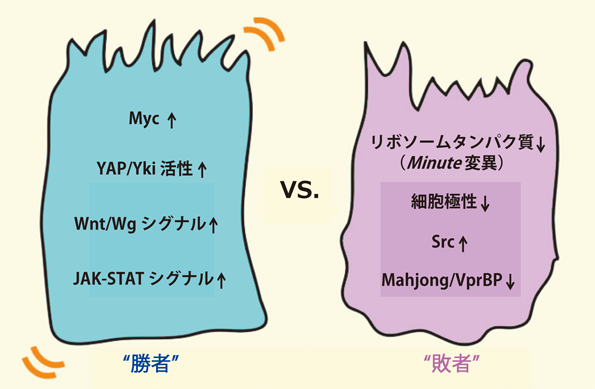
細胞競合が多くの研究分野に注目されるようになったのは,この現象がリボソームタンパク質遺伝子の変異に限られたものではなく,がん遺伝子mycの発現量の差によっても引き起こされることが示されたことによるだろう(図2).2004年,JohnstonとBaslerのグループはそれぞれ独立に,ショウジョウバエ翅成虫原基においてMyc発現量が相対的に高い細胞クローンが低い細胞クローンを駆逐していく現象を報告した11, 12).Mycの発現量が高い細胞クローンは細胞競合の常勝軍団として振る舞うことから「スーパーコンペティター」と名づけられた.Mycスーパーコンペティターによる周辺細胞の細胞死誘導のメカニズムとして,Morenoらは「リガンド捕獲モデル」に基づくJNK依存的細胞死であるとしたのに対し12),Johnstonらは細胞死遺伝子hidの発現誘導に起因することを示し,JNK依存的細胞死は主な要因ではないとしている11).興味深いことに,Johnstonらの最近の研究により,がん抑制遺伝子p53の機能が抑制された状況ではMyc高発現細胞は勝者から敗者に転じることが示されている13).
Mycタンパク質はリボソーム生合成を制御することで細胞の成長を促進することから,Myc細胞競合とMinute細胞競合の共通性が示唆される.一方で,Minute細胞競合では敗者の細胞死は勝者クローンとの境界上で起こるのに対し3, 5, 7),Myc細胞競合では勝者クローンから8細胞分以内の距離で離れた位置でも敗者の細胞死が起こるという相違点が示されている11).実際に,ショウジョウバエ培養細胞を用いたMyc細胞競合のin vitroモデル系において,勝者細胞から分泌された因子を介して離れた位置にいる敗者細胞が細胞死を起こすことが示されているが14),Morenoのグループはこの結果に異議を唱えている15).興味深いことに,マウスES細胞やエピブラストにおいてもMyc発現量の差による細胞競合が引き起こされることが報告され,哺乳類の発生初期において優良幹細胞を選別するのにMyc細胞競合が重要な役割を果たしている可能性が示された16, 17).このマウスにおけるMyc低発現細胞(敗者)の細胞死メカニズムとして,Myc高発現細胞(勝者)による貪食がその初期イベントであるとされている16).
がん遺伝子産物Mycを高発現する細胞がスーパーコンペティターとなることから,がんの発生・進展における細胞競合の役割が注目されるようになった.さらにこれを支持するように,がんに関わる種々のシグナル伝達経路の異常が細胞競合を駆動することが示されてきた.たとえば,Bakerらは大規模なショウジョウバエ遺伝学的スクリーニングを通じて,がん抑制経路Hippo経路の変異細胞[転写共役因子Yki(Yapホモログ)の活性が亢進した細胞]がスーパーコンペティターとなって周囲の野生型細胞を駆逐することを見いだした6).同様のHippo経路変異による細胞競合現象は,哺乳類培養細胞系においても観察されている18).興味深いことに,JohnstonらのグループはMycがYkiの転写ターゲットであることを示しており19, 20),Myc細胞競合とHippo経路変異による細胞競合との接点が示唆される.一方,VincentらのグループはWnt/Wgシグナルを亢進した細胞がスーパーコンペティターとして振る舞うこと21),またBachらのグループはJAK-STATシグナルを亢進した細胞が同じくスーパーコンペティターとなって周辺細胞を駆逐すること22)を,いずれもショウジョウバエ成虫原基において示した.これらWgシグナルおよびJAK-STATシグナル強度の差により引き起こされる細胞競合の敗者の細胞死は,いずれもMyc活性には依存しない.Wgシグナルの高い勝者細胞は,Wgシグナルの下流で発現誘導される分泌性のWgシグナル抑制因子Notumを周辺細胞に作用させて敗者の細胞死を引き起こすとされている21).一方,JAK-STATシグナルの高い細胞は,周辺の敗者細胞に細胞死遺伝子hid依存的な細胞死を引き起こすことが示されている22)(図2).
このように,種々のがん原性変異がスーパーコンペティターを生み出し,それらが少なくとも部分的には異なるメカニズムで周辺の正常細胞に細胞死を誘導して駆逐していくという現象が存在することがみえてきた.ただし,細胞増殖能を亢進させるようながん原性変異が必ずしもスーパーコンペティターを生み出すわけではない11).また興味深いことに,細胞周期を停止した組織においても細胞競合が起こることが報告されている23).今後これらの細胞競合メカニズムの詳細と生理的意義,またその普遍性と多様性を理解していくことが重要であると考えられる.
3)細胞競合によるがん抑制
がん制御における細胞競合の重要性を決定的に示したのは,細胞競合が上皮組織中のがん原性細胞の排除に働くという事実であろう.すなわち,細胞競合は細胞間相互作用を介した新しいタイプのがん抑制機構としても認識されるようになった.
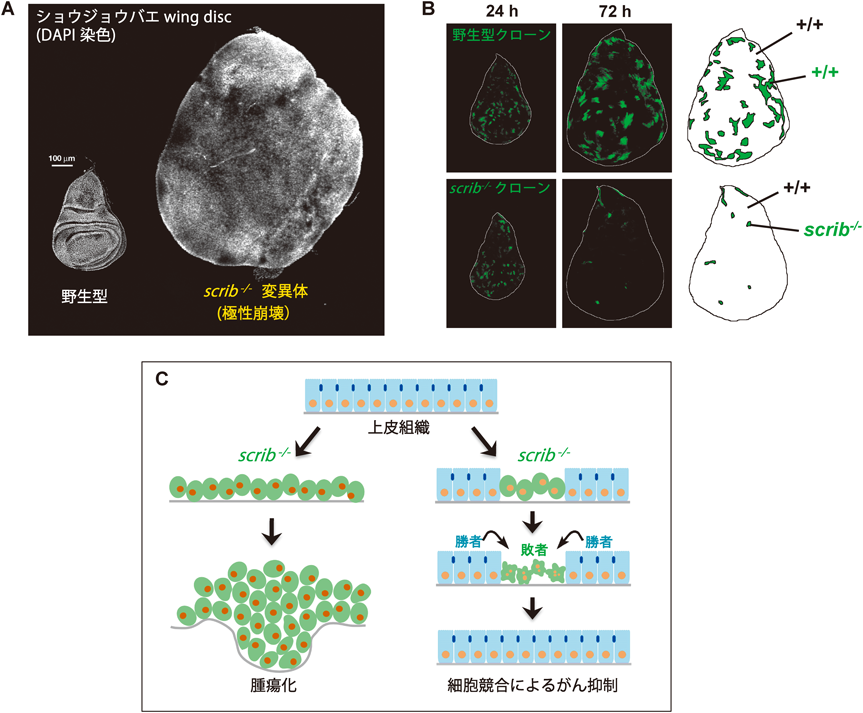
がんのほとんどは上皮由来であり,がんの発生・進展には上皮細胞のapico-basal極性の崩壊が密接に関わる24).ショウジョウバエ上皮においても同様に,apico-basal極性の崩壊は過剰な細胞増殖を引き起こす.たとえば,進化的に保存された極性遺伝子lethal giant larvae(lgl),discs large(dlg),scribble(scrib)などを欠損したショウジョウバエ変異体の成虫原基の上皮細胞は,大過剰に増殖して腫瘍を形成する(図3A).これらの変異組織は,極性の崩壊に伴って上皮構造が破壊され,かつ浸潤性を持ったヒトの進行がんに類似の特徴を示すことから,極性遺伝子lgl, dlg, scribは「neoplastic tumor suppressor」とも呼ばれる25).興味深いことに,これらneoplastic tumor suppressorの変異細胞を正常な成虫原基の一部にモザイク状に誘導すると過剰増殖せず,むしろ細胞死を起こして組織から排除されることがわかってきた26–29)(図3B).この事実は,apico-basal極性が崩壊した細胞は高い腫瘍原性を有するものの正常細胞に近接すると細胞競合の敗者となり,細胞死によって組織から排除されることを示唆している.筆者らは,scrib変異細胞を用いてがんの進展メカニズムを解析する過程でこの現象に気づき29),まさにMorataらが提唱した細胞競合の概念に合致する現象であることに衝撃を受けてその分子メカニズムの解析をスタートした.
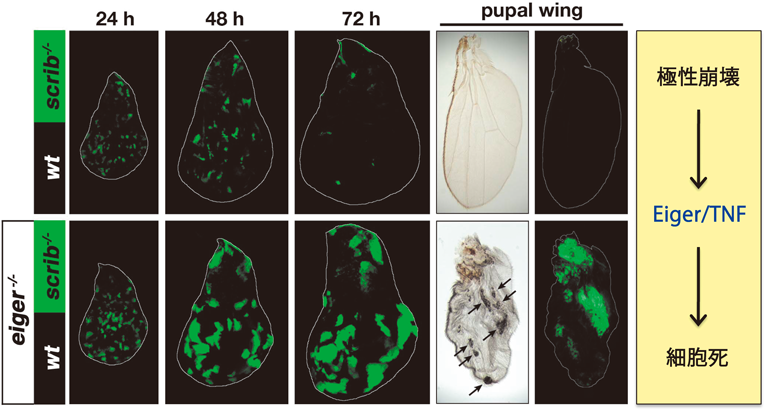
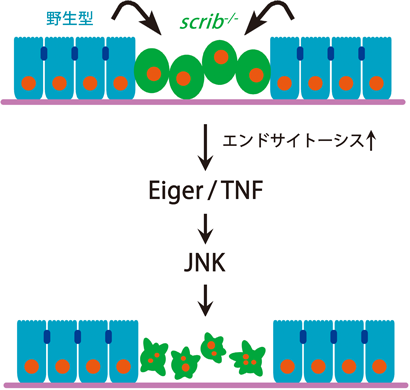
上述したように,ショウジョウバエ成虫原基の上皮組織全体でscrib遺伝子を欠損させると変異細胞は過剰に増殖して腫瘍を形成するが,遺伝的モザイク法を用いて組織中の一部にscrib変異細胞のクローンをモザイク状に誘導すると発生過程で排除される(図3C).筆者らはまず,マイクロアレイを用いて遺伝子発現プロファイル解析を行い,scrib変異細胞集団がJNKシグナルを活性化していることを見いだした(未発表).そこで,scrib変異細胞クローン内でJNKシグナルを阻害したところ,変異細胞クローンの排除が強く抑制されることがわかった(この現象はRichardsonらのグループによって最初に報告された28)).次に筆者らは,正常細胞に囲まれたscrib変異細胞内でのJNK活性化メカニズムを解析し,その上流でTNFホモログ分子Eigerが必須の役割を果たしていることを見いだした.Eigerはショウジョウバエで唯一のTNFスーパーファミリー分子であり,成虫原基で過剰発現させるとJNKシグナルの活性化を介して細胞死を誘導する.偶然にも,Eigerは筆者が大学院生時代に三浦正幸先生(現東京大学薬学系研究科教授)のもとで同定・命名した分子であった30).Eigerを欠損した成虫原基では,scrib変異細胞クローン内のJNK活性化が完全に阻害され,scrib変異細胞は排除されずに過剰に増殖して腫瘍を形成した(図4).さらに,Eiger-JNKシグナルの活性化メカニズムの解析を進め,scrib変異細胞内でエンドサイトーシス活性が顕著に亢進していることを見いだした.このエンドサイトーシス活性を阻害することでscrib変異細胞内のJNK活性化が抑制されること,またエンドサイトーシス経路の異所的な活性化がEiger-JNKシグナルの活性化に十分であること(未発表)から,scrib変異細胞内で亢進するエンドサイトーシス活性がEiger-JNKシグナルを活性化して細胞死を引き起こすと考えられた31)(図5).実際に,scrib変異細胞内でのJNK活性化は主にエンドソームで起こっており,このことはエンドソーム上にJNKシグナル活性化のためのアダプタータンパク質やスキャフォールドタンパク質が局在していることを示唆している.一方,scrib変異細胞がエンドサイトーシス活性を亢進させるメカニズムはいまのところ不明であるが,scrib変異により拡大した細胞のアピカル膜を減らすために起こる細胞応答である可能性が考えられる.
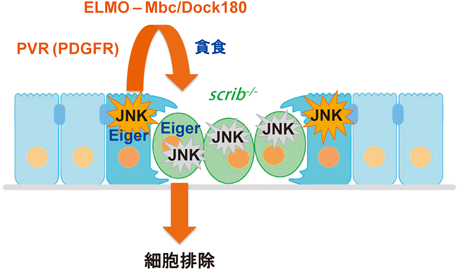
ここまでの解析のほとんどは,筆者がYale大学Tian Xu研究室にポスドクとして留学していた際に行ったものである.2007年に神戸大学医学研究科にて自身の研究室を主宰する機会を得て,この細胞競合現象の中で最も興味深い部分に取り組もうと考えた.すなわち,scrib変異細胞は周りを正常細胞に囲まれて初めて細胞死を起こして排除されるわけであるから,正常細胞側の役割を詳細に解析することでこの現象の全貌に迫れるのではないかと考えた.さらなる解析の過程で,興味深いことにscrib変異細胞に直接隣接する正常細胞(つまり勝者細胞)においても,JNKシグナルの活性化が起こっていることを見いだした.すなわち,正常な上皮細胞は隣に極性崩壊細胞が出現するとそれに応答してJNKシグナルを活性化すると考えられた.この正常細胞におけるJNK活性化は細胞死を誘導せず(極性を保っている上皮細胞はJNK誘導性細胞死に耐性を示す),むしろ隣接するscrib変異細胞の排除に促進的に働いていることがわかった.一方で,scrib変異細胞クローン内で観察される細胞死は,scrib変異細胞クローンと野生型細胞クローンの境界上に限局していることがわかった.つまり,scrib変異細胞集団の端から順に細胞死が起こっており,このことは正常細胞とscrib変異細胞間の直接的な接触が引き起こす細胞競合現象であることを示唆していた.そこで,勝者細胞においてJNKシグナルの下流で機能する分子を遺伝学的に探索したところ,JNKシグナルによりPDGF/VEGF受容体ホモログであるPVRが発現誘導され,さらにPVRシグナルの下流でアクチン骨格系を制御するELMO-Mbc/DOCK180経路を介して勝者細胞が貪食能を亢進し,近接する敗者細胞の排除を促進することを見いだした.勝者細胞において貪食機構を遺伝的に破綻させるとscrib変異細胞クローンのサイズが有意に増大したことから,勝者細胞による貪食は単なる死細胞のクリアランスではなく,積極的に細胞死誘導を行っていると考えられた32)(図6).これと類似の貪食による細胞死誘導として,エントーシスと呼ばれる細胞死形態が哺乳類の上皮細胞において知られており,実際にヒトのがん組織においてエントーシス様の細胞死が観察されることが報告されている33).
以上の筆者らの解析により,scribやdlgなどの変異により極性が崩壊したショウジョウバエ上皮細胞はエンドサイトーシス活性を亢進してEiger-JNK細胞死経路を活性化すること,またscrib変異細胞に隣接する正常細胞もJNKシグナルを活性化して貪食能を亢進し,隣のscrib変異細胞の排除を促進することがわかった.これら二つの細胞排除機構が同時に作用するのは極性崩壊細胞クローンと野生型細胞クローンの境界上の変異細胞のみであり,ゆえにクローン境界で特異的に変異細胞が排除されると解釈できる(図6).
筆者らは,これらの解析と並行して大規模なショウジョウバエ遺伝学的スクリーニングを実施し,極性崩壊が引き起こす細胞競合の最上流メカニズム,すなわち正常細胞と極性崩壊細胞がどのようにして互いのステータスの違い(あるいは細胞適応度ともいうべき状態指数)を認識するのかを理解しようと試みてきた.具体的には,scrib変異細胞クローンの周辺の正常細胞にのみEMS【(ethyl methanesulfonate)】誘導性の突然変異を誘導し,それによりscrib変異細胞クローンを排除できなくなるelimination-defective変異体(eld変異体),およびscrib変異細胞クローンの排除が顕著に亢進するsuper-eliminator変異体(sel変異体)を網羅的に探索した.約9000系統のショウジョウバエ変異体を樹立してスクリーニングを行い,多数のeld変異体およびsel変異体を単離することに成功した.その中でも特に強いelimination-defective表現型を示す複数の変異体の共通の責任遺伝子として,勝者細胞において敗者細胞の認識に関与すると考えられる細胞表面リガンドタンパク質Sasを同定した.さらにin vivo RNAiスクリーニングにより,敗者細胞側で機能するSasの受容体として受容体型チロシンホスファターゼPTP10Dを同定するとともに,このSas-PTP10Dシグナルが細胞競合を駆動するメカニズムを明らかにした(筆者ら,投稿中).興味深いことに,PTP10Dの哺乳類ホモログであるPTPRJはがん抑制遺伝子として機能することが報告されており34),ヒトのがんにおいても同様の細胞競合を介したがん抑制機構が存在する可能性が考えられる.現在筆者らは,Sas-PTP10Dシグナルを介した細胞競合の生理的役割を解析するとともに,哺乳類細胞系を用いてその普遍性の解析を進めている.
以上述べてきたような細胞間相互作用を介したがん原性細胞の排除現象が,哺乳類培養細胞系においても観察されることが藤田らのグループにより示されている.たとえば,がん遺伝子RasやSrcを活性化,あるいはscribをノックダウンしたイヌ腎臓由来MDCK細胞は,単独では生存して単層上皮細胞層を形成するが,その周囲を野生型MDCK細胞に囲まれると単層上皮細胞層から排除される35–37).ここで,scribノックダウン細胞はストレスキナーゼp38依存的に細胞死を起こして排除されるのに対し,RasやSrcを活性化した細胞は細胞死を起こすことなく主にアピカル側へと押し出されるように排除される.このような哺乳類培細胞系でみられる細胞排除とショウジョウバエ上皮でみられる細胞競合とを同一の現象として捉えるべきかどうかについては今後の研究の進展を待つ必要があるが,少なくともがん原性の変異細胞が正常細胞に囲まれると集団から排除されるというコンセプトは進化的に保存されているようである.では,実際の上皮がんは一体どのようにして極性崩壊細胞から生じうるのだろうか.興味深いことに,ショウジョウバエscribやdlgに変異を持つ極性崩壊細胞内でがん遺伝子Rasが活性化すると,変異細胞は細胞競合による排除を免れるだけでなく,むしろJNK依存的に高い増殖能と浸潤・転移能を獲得することが筆者らを含む複数のグループにより示されている28, 29, 38, 39).また,このようなscrib変異とRas活性化の協調によるがん化の促進は,哺乳類モデルにおいても起こることがHumbertらのグループにより示されている40–42).つまり,細胞競合による排除を免れるような第二のがん原性突然変異の獲得が極性崩壊細胞を競合の敗者から勝者へと転換し,がんの発生・進展を促すというシナリオの存在が考えられる.
がん制御においては,細胞間の競合のみならず協調機構が重要な役割を果たす.実際に,がんは単なる遺伝子変異の蓄積による細胞の異常増殖ではなく,遺伝子変異に起因するさまざまな細胞変化や細胞間相互作用が複合的に引き起こす「組織レベルのシステム破綻」であると近年捉えられるようになった.すなわち,がんの発生・進展メカニズムを理解する上で,がん微小環境における細胞間の協調メカニズムを生体レベルで理解することが重要である.ショウジョウバエのクローン解析技術を駆使した最近の研究により,がんの発生・進展に重要な役割を果たすと考えられる細胞間協調の新たなメカニズムがみえ始めている.
1)死にゆく細胞が引き起こす代償性増殖
1970年代半ば,Bryantらはショウジョウバエ成虫原基においてX線照射により大量の細胞死を誘導した際,生き残った細胞が余分に増殖することで組織サイズの恒常性を保つという興味深い現象を見いだした43).この発見から約30年を経た2004年,Steller, Hay, Morataのグループはそれぞれ独立にこの現象の解析を進め,組織中で死にゆく細胞が分泌性の増殖因子の発現を亢進することで代償的に周辺細胞の増殖が促されることを示し,「代償性増殖(compensatory cell proliferation)」のコンセプトが確立された44–46).これまでに,ショウジョウバエのみならず哺乳類などさまざまな生物種において代償性増殖が観察されることが報告され,創傷治癒や組織再生,さらには正常発生やがん制御への関与が示されている.実際に,ヒトのがん組織では細胞死が亢進していることが知られており47),がん原性細胞と死にゆく細胞との協調機構ががんの発生・進展に重要な役割を果たしている可能性が考えられる.
2)Srcによる細胞非自律的ながん進展
がんの悪性度に相関して,がん遺伝子Srcの活性や発現レベルが上昇することが知られている48).しかし,生体内でSrcを活性化した細胞がどのようにしてがんの発生・進展に寄与するのかについてはいまだ不明な点が多い.筆者らは,ショウジョウバエ複眼成虫原基の上皮組織において,ショウジョウバエSrcホモログ分子の一つSrc64Bを高発現する細胞クローンをモザイク状に誘導したところ,興味深い現象を見いだした.まず,正常細胞に囲まれたSrc活性化細胞クローンは過剰増殖せず,むしろ細胞死を起こして組織から排除されることがわかった.これは,Src活性化細胞が正常細胞に対峙すると細胞競合の敗者になることを示している.さらに興味深いことに,Src活性化細胞クローンは自身は排除されつつも周囲の正常細胞の増殖を強く促進し,複眼組織に腫瘍を形成させた.この細胞非自律的な増殖促進機構を解析した結果,Src活性化細胞は細胞内F-actinを過剰に集積してHippo経路の不活化を介してYkiを活性化し,同時にSrc活性化細胞内で活性化したJNKシグナルがYki活性を周辺細胞に伝播させることで周辺細胞の過剰増殖が起こることがわかった49).このJNK依存的なYki活性の伝播のメカニズムは現在のところ不明であるが,成虫原基における創傷治癒や組織再生の過程でJNK活性の伝播50)や細胞非自律的なYki活性化51)が認められており,それらとの関連も含めて非常に興味深い.また,Src活性化細胞が自身ではなく周辺細胞の腫瘍化を促進するという事実は,細胞間協調を介したがん進展におけるSrc活性の重要性を示唆している.筆者らは,さまざまながん遺伝子の活性を亢進させた細胞集団をSrc活性化細胞クローンに隣接するように誘導し,細胞間協調によりがん進展を引き起こすがん遺伝子活性をスクリーニングした.その結果,Rasを活性化した細胞集団がSrc活性化細胞集団の近傍に生じると,細胞間コミュニケーションを介して双方の細胞集団が相利共生の悪性腫瘍を形成することを見いだした(筆者ら,投稿準備中).ショウジョウバエ遺伝学を用いた今後の解析により,がん遺伝子活性の組織内不均一がどのようにしてがん進展を引き起こすのか,その基本原理が明らかになっていくと期待される.
3)がん原性炎症による細胞非自律的ながん進展
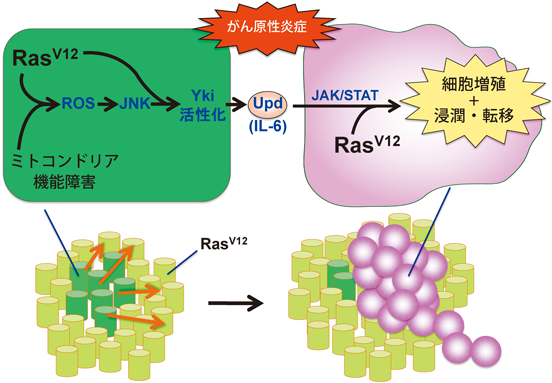
ショウジョウバエ成虫原基においてRasを活性化した細胞クローンをモザイク状に誘導すると,これらの変異細胞は過剰に増殖して良性腫瘍を形成する.筆者らは,このRas活性化細胞クローンにさらなる突然変異を導入し,その周辺細胞の増殖能が変化するような変異を探索した.その結果,Ras活性化細胞内でミトコンドリアの機能障害を起こすような第二の突然変異が誘導されると,その周辺細胞が大過剰に増殖することがわかった.そのメカニズムとして,Ras活性化とミトコンドリア機能障害が同時に起こると活性酸素種(ROS)が大量に産生され,これによりJNKシグナルが活性化すること,さらにこのJNKシグナルとRasシグナルが協調して細胞内F-actinの集積を引き起こすことでYkiが活性化し,その転写ターゲットである炎症性サイトカインUnpaired(Upd:IL-6ホモログ)や分泌性増殖因子Wnt/Wgが発現誘導されることが明らかとなった(図7).これらの分泌性増殖因子を受け取った周辺細胞が過剰に増殖する一方で,ミトコンドリア機能障害を起こした変異細胞はむしろ細胞周期を停止することがわかった.ミトコンドリア機能障害は,多くのヒトのがんで高頻度に認められる細胞変化である.がんにおけるミトコンドリア機能障害は,ミトコンドリアDNAの体細胞突然変異を始めとした種々の偶発的要因によって引き起こされると考えられるため,実際のヒトのがんにおいては,Rasを活性化した腫瘍組織の一部でミトコンドリア機能障害が誘発されると想定される.そこで,そのような状況をショウジョウバエ複眼原基において再現したところ,ミトコンドリア機能障害を起こしたRas活性化細胞から産生されたUpdが周辺の(正常なミトコンドリア機能を持つ)Ras活性化細胞のJAK-STATシグナルを活性化し,その結果周辺の良性腫瘍が悪性化する(浸潤・転移能を獲得する)という劇的な現象が引き起こされることがわかった52)(図7).
本スクリーニングにおいては,ミトコンドリア機能障害を引き起こす一連の突然変異の他にも,炎症性サイトカインUpdを介した細胞非自律的増殖を引き起こす突然変異が多数単離された.その中の一つに,エンドソームの生成に必要なRab5遺伝子の変異も含まれていた.Rab5遺伝子を欠損した細胞集団は,細胞表面受容体であるTNF/EigerおよびEGFRのエンドサイトーシスを介した分解が阻害され,それにより活性化されたJNKおよびRasシグナルが協調してF-actinの集積を介してYkiを活性化し,Updの発現を誘導することがわかった53).エンドサイトーシス経路の破綻もヒトのがん組織でみられる共通の細胞変化の一つであり,細胞間協調を介したがん進展におけるエンドサイトーシス破綻の重要性が示唆される.また,このような炎症性サイトカイン産生を介したがん進展がさまざまな突然変異により引き起こされることがみえてきたことから,筆者らはこの現象を「がん原性炎症(oncogenic inflammation)」と呼び,その統一的理解を目指している54, 55)(図7).
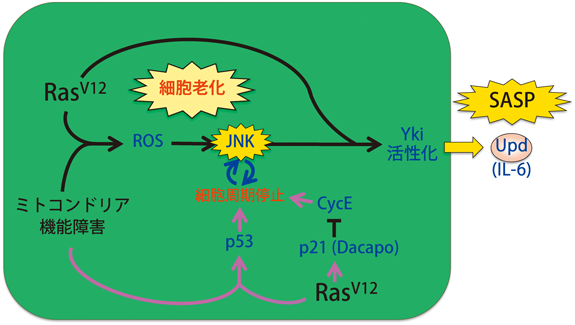
さて,Ras活性化とミトコンドリア機能障害によってがん原性炎症を起こした細胞を詳細に解析したところ,この変異細胞集団は細胞死に対して耐性を示すとともに,細胞周期を停止して顕著に肥大化していることがわかった.さらに興味深いことに,これらの変異細胞はSA-β-gal(senescence-associated β-gal)活性の上昇,Cdk阻害タンパク質Dacapo(p21/p27ホモログ)の発現上昇,ヘテロクロマチン化,DNA損傷応答など,一連の細胞老化の表現型を呈することがわかった.このことは,がん原性炎症における炎症性サイトカイン産生が,細胞老化に随伴して起こる分泌タンパク質の高発現現象SASP(senescence-associated secretory phenotype)であることを意味している.またこの観察事実は,細胞老化およびそれに伴うSASP現象が無脊椎動物においても進化的に保存されていることを初めて示すものとなった.さらなる解析により,Rasシグナルの活性化により発現上昇するDacapo,およびRas活性化とミトコンドリア機能障害の協調により発現誘導されるp53が細胞周期を停止させ,さらにこの細胞周期停止とJNK活性化がポジティブフィードバックループを形成してJNK活性化をブーストし,持続的な炎症性サイトカイン産生を実現していることがわかった56)(図8).
以上のように,Rasシグナルを活性化した細胞においてある特定の突然変異/細胞変化が起こると,その周辺細胞の増殖が劇的に亢進するという現象の存在がみえてきた.このような強烈な生命現象が存在する意義は何であろうか.この問いに答えるため,筆者らは現在,生理的に起こるがん原性炎症の探索と正常発生過程における細胞老化(プログラム細胞老化)の解析を進めている.
本稿では,ショウジョウバエがん制御モデルにおける上皮細胞間の競合・協調現象とそのメカニズムについて,筆者らの最近の研究成果を中心に概説した.細胞間の競合と協調という対照的な現象をそれぞれ個別に紹介したが,実は生体内ではこれら二つの現象は常に同時に起こっているということを忘れてはならない.たとえば,ショウジョウバエ成虫原基において細胞競合の敗者となった細胞は,近接する勝者細胞の増殖を代償性増殖により促進すると考えられる.また,Srcを活性化した細胞やRab5を欠損した細胞は,それぞれ異なるメカニズムで周辺細胞の増殖を強く促進しつつ,自身は細胞競合の敗者となって組織から排除されていく.少なくともショウジョウバエの上皮においては,細胞競合と細胞間協調はいずれも似たような細胞内シグナル伝達経路を状況依存的に使い分け,そのアウトプット(細胞変化)を導いているようにみえる.つまり,細胞内のシグナルクロストークと細胞間のコミュニケーションという二つの階層のネットワークの総和が組織としての振る舞いを決定しており,これを包括的に理解するには競合と協調のそれぞれの分子基盤を詳細に理解する必要がある.
1970年代にショウジョウバエで発見され2000年代に確立された「細胞競合」と「代償性増殖」という二つの魅力的なコンセプトは,いまやさまざまな生物種に適用され,その解析は数理モデルを用いた理論的アプローチにまで広がりつつある57).一方で,先述したように細胞競合の分子メカニズムについてはいまだ論争も多く,定まった共通認識はまだ限られている.これは,トリガーの違いによって異なるメカニズムの細胞競合が引き起こされるという事実にも起因している.筆者らは,生体内で細胞競合を引き起こしうる遺伝子変異の網羅的探索・同定も進めており,その共通項から細胞競合現象の本質に迫ることができないかと考えている.細胞間の競合・協調現象を通じて,新たな生物学的コンセプトが生まれてくることを期待している.
追記:2016年10月26日,Gines Morata博士の呼びかけにより各国の細胞競合の中心研究者がマドリッドに集結し,Peter Lawrence博士の立会いのもと,細胞競合現象の定義付けについての議論がなされた(下の写真).現時点では,1)Context dependency(状況依存的に勝者と敗者が決まり,敗者が排除される),2)Short range(直接接触あるいは数細胞の距離で起こる相互作用)の2つの特徴を有するという点でコンセンサスが確認された.