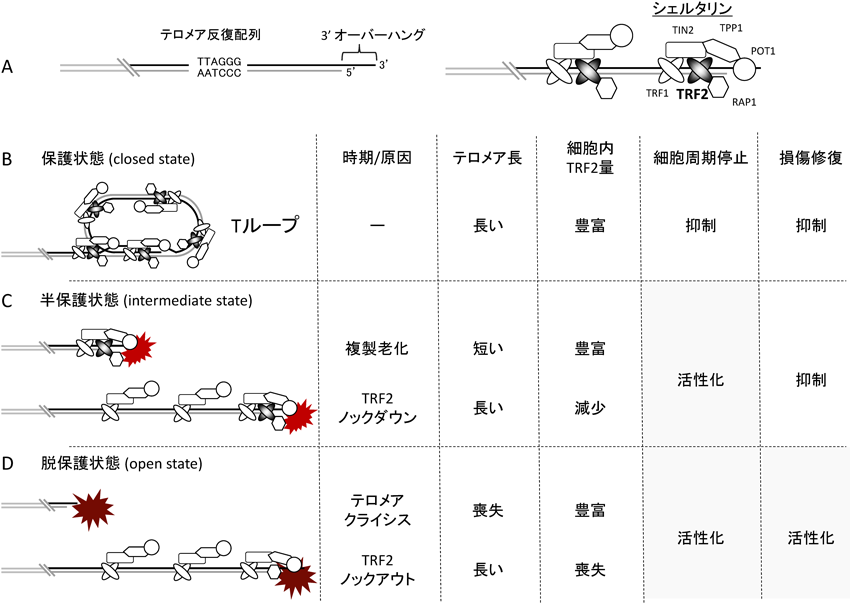
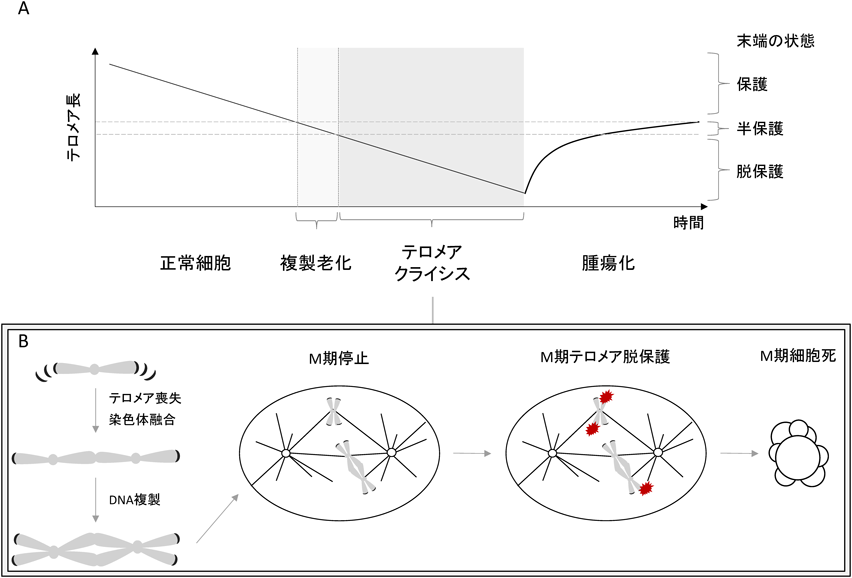
テロメアクライシス期におけるM期停止を介した細胞死機構Mechanism of mitotic cell death during telomere crisis
京都大学白眉センター/大学院生命科学研究科The Hakubi Center for Advanced Research/Graduate School of Biostudies, Kyoto University ◇ 京都府京都市左京区吉田近衛町 ◇ Yoshida Konoe-cho, Sakyo-ku, Kyoto, Japan
発行日:2016年12月25日Published: December 25, 2016