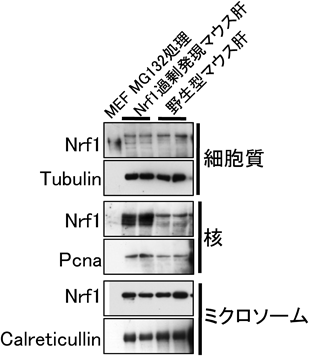
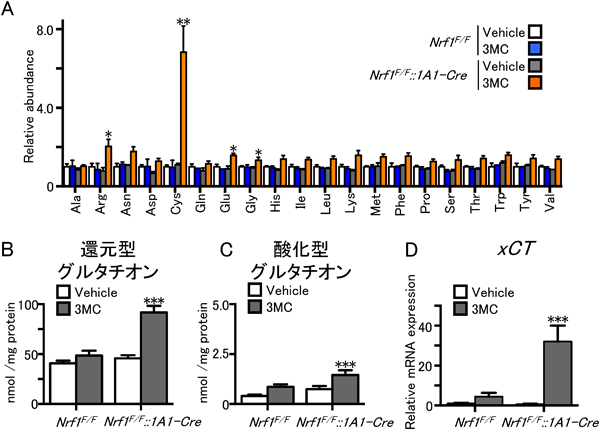
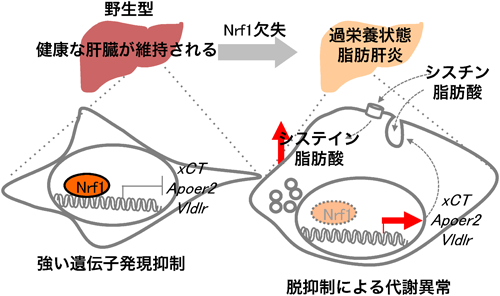
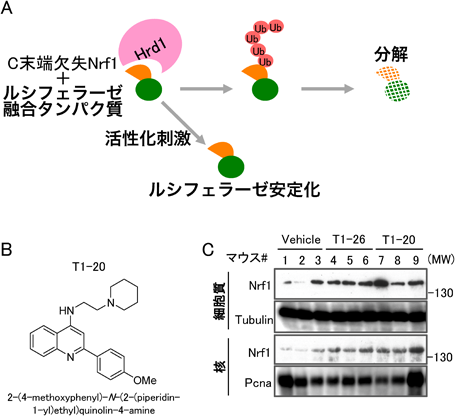
Nrf1(NFE2L1)の転写抑制能による細胞内チオール量および脂肪酸代謝制御Intracellular thiol contents and fatty acid metabolism pathways are regulated by the Nrf1 (NFE2L1) with its transcriptional suppressor activity
佐賀大学農学部生命機能科学科生化学分野Laboratory of Biochemistry, Department of Applied Biochemistry and Food Science Faculty of Agriculture, Saga University ◇ 〒840‒8502 佐賀県佐賀市本庄町1番地 ◇ 1 Honjyou-machi, Saga 840–8502, Japan
発行日:2016年12月25日Published: December 25, 2016