アルギニンに富む膜透過ペプチドの細胞内移行Membrane translocation of arginine-rich cell-penetrating peptides
京都大学化学研究所Institute for Chemical Research, Kyoto University ◇ 〒611–0011 宇治市五ケ庄 ◇ Gokasho, Uji, Kyoto 611–0011, Japan
発行日:2017年2月25日Published: February 25, 2017
アルギニンに富む膜透過ペプチド(アルギニンペプチド)との架橋体あるいは複合体形成により,タンパク質をはじめとする種々の細胞膜不透過性の生理活性分子を細胞内に送達できることが知られている.細胞表面のプロテオグリカン等との相互作用により,アルギニンペプチドは細胞表面に引き寄せられ,その後,エンドサイトーシスと形質膜の直接透過の二つの経路を使い分けることにより,細胞内に移行する.また,この取り込み経路はアルギニンペプチドとの架橋体/複合体の物性や濃度などの条件によって変化する.アルギニンペプチドのエンドサイトーシスによる細胞内移行には,アクチン駆動性の液相エンドサイトーシス(マクロピノサイトーシス)も関与する.形質膜の直接透過には膜電位が重要な役割を持ち,ピレンブチレートなどの対イオン分子を介在させることで膜透過が促進される.
© 2017 公益社団法人日本生化学会© 2017 The Japanese Biochemical Society
近年,cell-penetrating peptide(CPP)と総称される膜透過性ペプチドを用いた細胞内への生理活性物質導入法がしばしば用いられるようになってきた1–4).この方法は,CPPと共有結合(化学結合)で架橋したり,非共有結合的な安定複合体を形成させたりすることによって,通常なら細胞膜を透過できない高分子量薬物やナノ粒子を細胞内へ効率的に送達できる.タンパク質の細胞内導入においては,CPPとの融合タンパク質を用いることも可能である.手法的な容易さと細胞レベルでの結果の得られやすさから,CPPの応用例は年々増加している4).
CPPの細胞内への移行は,エンドサイトーシスと形質膜の直接透過の二つの観点から説明される5–7).前者は細胞の生理的な取り込み機序という観点から,後者は生物物理化学的観点からの説明といえる.CPPと細胞内に導入する物質との架橋体・複合体の物性と細胞側の状態により,CPPを用いた細胞内への送達は,エンドサイトーシスまたは直接膜透過を介した経路の複合的な兼ね合いにより行われる.
代表的なCPPとして,HIV-1のTatタンパク質由来の塩基性ペプチド8)(本稿ではTATと記載する:GRKKRRQRRRPPQ)やオクタアルギニン(R8:RRRRRRRR)などのオリゴアルギニンならびに類縁体9, 10),ショウジョウバエのアンテナペディアホメオドメインタンパク質由来の塩基性ヘリックスペプチド(penetratin:RQIKIWFQNRRMKWKK)11),神経ペプチドガラニンから派生した誘導体TP10(AGYLLGKINLKALAALAKKIL-アミド)12)などがあげられる.これらのペプチドは共通して細胞膜の透過性と細胞内への薬物などの導入能を有すると考えられるが,細胞内への移行機序はそれぞれのペプチドにより少しずつ異なると考えてよい.
TATやR8などのオリゴアルギニンは塩基性アミノ酸であるアルギニンに富む構造的特徴を有している.このアルギニンに富むCPPは,その高い細胞内移行効率により,最も汎用されるタイプのCPPの一つである.これらを用いて,タンパク質・ペプチド,リポソーム,ナノ粒子などさまざまな薬物や物質が細胞内に導入され,細胞内で所望の活性を発揮したことが報告されている6, 7).in vivoレベルでの応用に関しては今後の検討を待つ部分も多いが,細胞レベルの実験においては,試みるべき手法の一つとなっている.筆者らは,このように塩基性の高いペプチドがなぜ効率的に細胞内に移行できるのかに関して興味を持ち,研究を進めてきた.本稿では,エンドサイトーシス,形質膜の直接透過の両面からアルギニンペプチドの細胞内移行に関して筆者らの知見をもとに概説する.
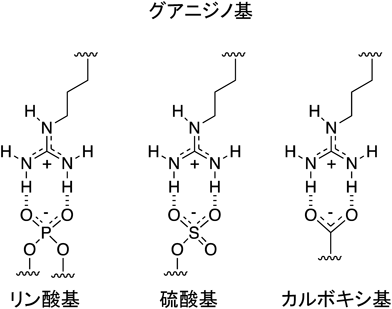
アルギニンはグアニジノ基(pKa~12.5)を側鎖に有している.グアニジノ基は硫酸基,リン酸基,カルボキシ基と静電的相互作用を行うとともに,二つの水素結合を形成可能である(図1).アルギニンペプチドが顕著な細胞内移行性を示すには,一般に,分子内におおむね6個あるいはそれ以上のアルギニンが含まれていることが重要である9).同じく塩基性アミノ酸で側鎖にアミノ基(pKa~10.5)を有するリシンのオリゴマーも細胞内移行性を示さないわけではないものの,同数のアルギニンとリシンを有するペプチドの場合,前者は後者に比べて数倍高い細胞内移行性を有することが多い13).アルギニンペプチドが高い細胞内移行性を示す大きな理由として,筆者らは細胞表面への吸着性を考えている.エンドサイトーシス,形質膜の直接透過のいずれの経路で細胞内に取り込まれる場合も,まず,細胞表面付近にアルギニンペプチドが到達する必要がある(図2).アルギニンペプチドは細胞表面のプロテオグリカン(硫酸化された糖鎖で修飾されたタンパク質)との相互作用により,効果的に細胞表面に移行・濃縮される.この際,プロテオグリカンの硫酸基とグアニジノ基の間の水素結合形成が重要と考えられる.水素結合,静電相互作用ともに水中では比較的弱い相互作用であるため,効果的なプロテオグリカンとの相互作用には6個程度あるいはそれ以上のアルギニンが必要となる.また,リシンは,これらの官能基と相互作用しても水素結合を一つしか形成できないため,同数のアルギニンとリシンのオリゴマーでは前者の細胞表面との相互作用が後者に比べてより強いと考えることで,リシンに対するアルギニンの優位性が説明できる.アルギニンの塩基性と水素結合形成能のいずれが細胞膜との相互作用ならびに細胞内移行により重要かという点に関しては,Rothbardらが興味深い成果を報告している14).同グループはグアニジノ基の水素をメチル基に置換した誘導体を合成し,細胞内への移行性を検討した.メチル化したグアニジノ基は未修飾のグアニジノ基より高い塩基性を示すにも関わらず,メチル化されたものに比べて,未修飾のアルギニンのオリゴマーが細胞内に数倍多く取り込まれた.また,同グループは,オリゴアルギニンのオクタノールと水の間での分配を調べたところ,単独ではまったくオクタノール相に移行しなかったオリゴアルギニンが,カルボキシ基を有する脂肪酸(ラウリン酸)を添加することによりオクタノール相に分配したことを示している.この結果は,疎水性の対イオン分子と相互作用することにより,アルギニンペプチドが疎水性の膜環境に移行しうることを示唆するものである.同論文ではアルギニンペプチドの細胞内移行における膜電位の重要性に関しても指摘されている.通常,細胞内の電位は細胞外に比べて低く保たれており,ポリカチオンであるアルギニンペプチドが電位差を駆動力として電位の低い方向,つまり細胞内に流入するというものである14).
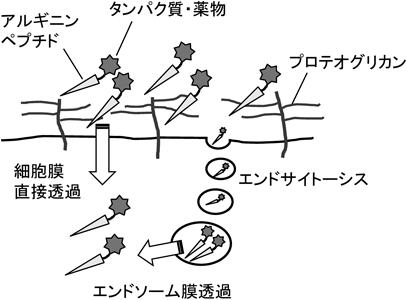
アルギニンペプチドの細胞内移行における細胞表面のプロテオグリカンの重要性に関しては,筆者を含むさまざまな研究グループが確認している15–20).アルギニンペプチドとヘパリンなどの硫酸化糖鎖はサブµM程度の解離定数で複合体を形成することが報告されている.また,細胞表面の硫酸化糖鎖を欠失した細胞株では,アルギニンペプチドの細胞内への移行は,野生株の1/10程度まで低下する16).
前述のようにアルギニンペプチドとその細胞内導入物質(カーゴ)との架橋体・複合体の細胞膜透過は形質膜の直接透過とエンドサイトーシスの間の兼ね合いによって行われる21).通常は,エンドサイトーシスで細胞内に取り込まれる経路が主であるが,細胞表面へのアルギニンペプチドの集積度が高く,カーゴが比較的小さい場合(条件にもよるが,分子量にして2000~3000程度),直接膜透過がみられる.細胞表面へのアルギニンペプチドの集積度を高める要件として,投与時のペプチド濃度を高める,アルギニンペプチド中のアルギニン数を多くする,アルギニンペプチドと相互作用する血清タンパク質の非存在下に細胞とアルギニンペプチドをインキュベーションする,などが考えられる22).アルギニンペプチド自体がどのようにして形質膜を直接透過するかに関しては不明な点が多いが,アルギニンペプチド自体が膜透過性を有していても,これと連結された(あるいは複合体を形成する)カーゴは一般に膜透過性を有しない.したがって,このサイズが大きくなるに従い,架橋体あるいは複合体全体の物性に与えるカーゴ分子の影響が大きくなり,膜透過性が低下すると考えられる23).
前述のように,アルギニンペプチドの直接膜透過においては,アルギニンペプチドが細胞表面に高濃度で集積することが重要である.ここで,エンドサイトーシスにより,アルギニンペプチドが細胞内部に取り込まれると,細胞表面へのアルギニンペプチドの集積度が低下することになり,結果的に直接膜透過は抑制される.アルギニンペプチドおよびその架橋体/複合体が細胞内へ主にエンドサイトーシスにより取り込まれる理由の一つはこれである.エンドサイトーシスはATP駆動型の取り込み機構であり,低温で働きを休止する22).このような低温処理条件下に細胞とアルギニンペプチドとをインキュベーションすると,アルギニンペプチドの形質膜からの直接透過による細胞内へのペプチドの総取り込み量は,37°Cのときのそれより多いという例も起こりうる24).エンドサイトーシスと直接膜透過はこのようなバランスの上に成り立っていると考えられる.
TATなどのアルギニンペプチドの膜透過性が報告された直後は,アルギニンペプチドの膜透過はエンドサイトーシス等の生理的取り込み経路を介しない,未知の機序による直接透過と考えられていた8).しかし,その後の検討により,細胞移行を観察する際の細胞固定操作によりペプチドの細胞内局在が大きく変わることがわかり,上述のように条件を選ぶことにより直接膜透過は可能であるものの,アルギニンペプチドの細胞移行にはエンドサイトーシスが大きく関与することが明らかとなった25, 26).
アルギニンペプチドの細胞内へのエンドサイトーシスによる取り込みにおいて特徴的な経路としてマクロピノサイトーシスがあげられる6, 18, 27, 28).マクロピノサイトーシスはアクチン駆動性の液相エンドサイトーシスである29).マクロファージなど一部の細胞を除いた多くの細胞においては,マクロピノサイトーシスは常時稼働しているのではなく,細胞表面での何らかの刺激を感知して誘導されることが知られている29).アルギニンペプチドと細胞表面との相互作用によりアクチンの再構成が誘起され,これが細胞膜の隆起を促し,膜の波打ち(ラフリング)状態を誘導する.これに続く膜融合により周囲の溶質や細胞表面に付着した物質を内部に取り込んだ脂質小胞(マクロピノソーム)が形成される.上述のようにアルギニンペプチドは細胞表面に強く吸着する特性を有するが,細胞表面に吸着されたアルギニンペプチドは結果的にマクロピノソームに高効率で取り込まれ,細胞内に移送される18).マクロピノサイトーシスの他,アルギニンペプチドがクラスリン依存性エンドサイトーシスやカベオラ依存性エンドサイトーシスで細胞内に取り込まれることも報告されている30).アルギニンペプチドのクラスリン依存性エンドサイトーシスには膜結合型プロテオグリカンの一種であるシンデカン-4が取り込み受容体として機能することを筆者らは最近見いだしたが,これに関しては後述する31).
マクロピノサイトーシスの特徴として,その名前の示すとおり,クラスリン依存性エンドサイトーシスやカベオラ依存性エンドサイトーシスにおける取り込み脂質小胞(エンドソーム)に比べて,大きなサイズの小胞を形成することにある32).クラスリン依存性エンドサイトーシスやカベオラ依存性エンドサイトーシスはエンドソームがクラスリンやカベオリン-1といったコートタンパク質に覆われる形で形成されるため直径が100 nm程度に規定されるのに対し,マクロピノサイトーシスはアクチンの重合により誘起されるので,マクロピノソームの大きさはタンパク質により規定されないことがその理由として考えられ32),このため,マクロピノソームの直径は数百nmから数µmにも達するとの報告もある33, 34).したがって,ナノサイズの大きな分子や粒子の細胞内への取り込みには,クラスリンやカベオラ依存性エンドサイトーシスよりも,エンドソームサイズの大きいこの経路が優先的に使われる可能性もある.近年,ウイルスの宿主細胞内への侵入にもこの経路がしばしば利用されていることが報告され35),その意味からもマクロピノサイトーシス経路が注目されている.
アルギニンペプチドの細胞内移行におけるマクロピノサイトーシスの関与に関しては,筆者らおよび他のグループも報告している.アルギニンペプチドと細胞表面のプロテオグリカンとの相互作用により,何らかのシグナルが細胞内に伝達され,これが細胞内のプロテインキナーゼCαやRac-1タンパク質を活性化し,アクチンの再構成を経て,膜の波打ち(ラフリング)状態とマクロピノサイトーシスが誘導されることを筆者らは報告している27, 36).細胞表面のプロテオグリカンが欠失した細胞ではアルギニンペプチドの細胞内への取り込みが大きく減少するとともに,アクチンの再構成やマクロピノサイトーシスの誘導がみられにくくなる16).
アルギニンペプチドの細胞内取り込みにおけるエンドサイトーシスの関与が示唆されたが,取り込みに関与する受容体が存在するかということが次の疑問として出てくる.これらの受容体が同定できれば,アルギニンペプチドやその類縁分子の細胞内取り込み機序が明らかにでき,より効率的な細胞内送達系が開発できる可能性がある.アルギニンペプチドの細胞内取り込みにおけるプロテオグリカンの寄与について上述したが,プロテオグリカン自体が受容体として働く可能性に加え,たとえば線維芽細胞成長因子(FGF)-2の場合にみられるように,プロテオグリカンとの相互作用により真の受容体との結合親和性が高められ,受容体の活性化が誘起される可能性も考えられる37).アルギニンペプチドと関連するタンパク質やペプチドの受容体として,たとえばHIV-1 Tatの全長タンパク質(86アミノ酸)の細胞内への取り込みには血管内皮増殖因子(VEGF)受容体(VEGFR2)が関与することが報告されている38).一方,Tatタンパク質の48~60位に対応する塩基性配列が代表的なアルギニンペプチドの一つであるTATであるが,TATの細胞内取り込みにVEGF受容体は関与しない.Tat全長タンパク質はケモカイン受容体CXCR4と結合し,HIV-1感染に対するアンタゴニストとして働くが,これはその11~50位に対応するシステインに富む領域を介して行われるもので,TATペプチドに対応する塩基性領域はCXCR4との相互作用には関与しない39, 40).
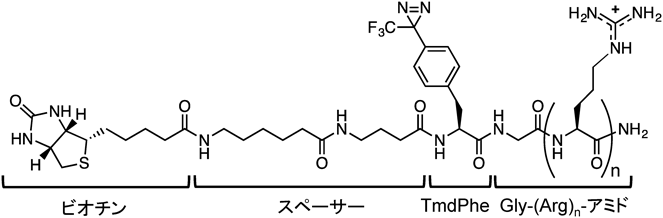
筆者らは,富山大学の畑中らとの共同研究により,ジアジリン誘導体を用いた光架橋によりアルギニンペプチドの受容体の同定を試みた41).アルギニンペプチドの一つ,ドデカアルギニン(R12)はR8よりも高い細胞表面への吸着性と細胞内移行性を有する.これにトリフルオロメチルジアジリンのフェニルアラニン誘導体42)と架橋産物の単離のためのビオチンを連結した誘導体(図3)を合成した.この光親和性プローブと細胞膜上の受容体との相互作用を期待して,37°Cで30秒処理したのち,4°C条件下で紫外線を3分間照射した.一方ネガティブコントロールとして細胞膜透過性をほとんど示さないテトラアルギニンR4をR12の代わりに配置したペプチドも調製した.紫外線照射後,細胞を溶解し,ストレプトアビジンビーズで架橋産物を単離し,SDS-PAGEに処した.R12特異的なバンドが高分子量領域に認められたので,このバンドを切り取り,プロテアーゼ処理後,質量分析を行ったところ,バンドは細胞骨格タンパク質であるミオシン-9に対応することが示唆された.ミオシン-9はCXCR4と共局在するという報告があったことから43),CXCR4をsiRNAを用いてノックダウンしたところR12の細胞内への取り込みは約30%低下した.また,CXCR4はGiタンパク質と共役しており,このシグナル伝達阻害剤である百日咳毒素(PTX)存在下でもR12の取り込みが約30%減少した.R12とCXCR4の細胞表面での共局在が認められるとともに,マクロピノソームマーカーである70 kDaデキストラン,CXCR4, R12が共局在を保ったまま細胞内に移行することも確認された.これにより,CXCR4はR12と相互作用することでマクロピノサイトーシスを誘導することがわかった.興味あることに,R12より鎖長の短いR8やTATの細胞内への取り込みに関しては,CXCR4のノックダウンやPTX処理の影響はみられなかった.この結果は,アルギニンペプチドの中でも複数のマクロピノサイトーシス(あるいはエンドサイトーシス)による取り込み経路が存在することを示したものである.
CXCR4とそのリガンドであるSDF-1(CXCL12)との相互作用によりアクチンの再構成が生じることが報告されていた44).筆者らは,この際マクロピノサイトーシスも誘導されるのではないかと考えた.実際に,SDF-1添加時にマクロピノサイトーシスのマーカー分子である70 kDaデキストランの細胞内への取り込みが大きく亢進することを見いだした.また,CXCR4はCD4を介するHIV-1の宿主への感染時の補助受容体として働くことが知られているが,HIV-1の表面タンパク質であるgp120を細胞に投与してもマクロピノサイトーシスが誘起されることがわかった.つまり,CXCR4はそのリガンドと相互作用することにより,細胞にマクロピノサイトーシスを誘起する受容体であることが示された.HIV-1は細胞表面における膜融合により宿主細胞に感染することが知られている.ところが一方,gp120とCXCR4の相互作用はCXCR4の内在化を誘導する.HIV-1の宿主細胞への感染は,CXCR4との相互作用による膜融合・感染の進展と受容体の内在化による感染阻害の二つのバランスの上に成り立っている可能性がこの結果から示唆された.
次に筆者らはR8にトリフルオロメチルジアジリンのフェニルアラニン誘導体と架橋産物の単離のためのビオチンを連結した誘導体(図3)を合成し,R8の細胞内取り込み受容体を探索した.この結果,細胞質に存在するとされるタンパク質lanthionine synthetase component C-like protein 1(LanCL1)が架橋産物として得られた45).LanCL1の細胞内での役割については不明の点が多いが,LanCL1を過剰発現させた細胞においては,R8の細胞内への取り込みが亢進した.このことから,LanCL1は何らかの機序によりR8の細胞内への取り込みを促進している,あるいは細胞内に移行したR8はLanCL1と相互作用することによりその取り込みをいっそう促進する可能性も考えられる.しかし,これに関しては今後の検討が必要である.
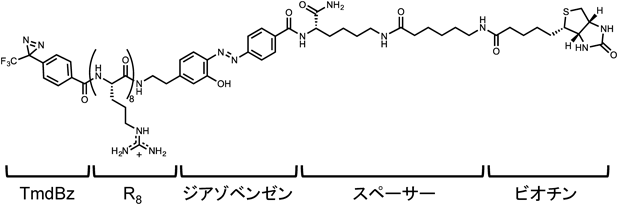
光架橋を用いた上記の二つの実験で問題となったのは,架橋体を単離する際に用いたストレプトアビジンビーズへの,他のタンパク質の非特異的吸着である.また,目的の受容体は細胞膜に存在すると考えられ,単離の際に膜タンパク質を効果的に回収することができれば,LanCL1のように細胞質あるいは核由来のタンパク質の混入を避けることができ,単離が容易になることが考えられる.これらを考慮して,筆者らはNa2S2O4処理により選択的に切断可能なジアゾベンゼン46, 47)をビオチンと光架橋団とR8の間に持つ,光架橋プローブ(図4)を開発した.光架橋後に細胞を溶解し,ストレプトアビジンビーズを用いて目的タンパク質を濃縮した後,Na2S2O4処理により架橋体を選択的にビーズから単離することができれば,非特異的に吸着された夾雑タンパク質はビーズにそのまま保持されるので,目的物の解析が大きく加速できると期待される.この光架橋プローブと,細胞溶解液の膜画分を選択的に回収する操作を組み合わせることで,バックグラウンドとなる夾雑タンパク質は劇的に低減され,このアプローチの実用性が確認された.SDS-PAGEにおいて,R8と処理した細胞に特異的な七つのバンドが認められ,これらをトリプシン消化し質量分析により解析したところ,17種類のタンパク質が候補として得られた.これらのタンパク質をsiRNAによりノックダウンしたところ,シンデカン-4をノックダウンした細胞においてのみR8の細胞内取り込みが抑制された.詳細な検討の結果,シンデカン-4はクラスリン依存性エンドサイトーシスによるR8の細胞内への取り込みに関与する受容体であることが示唆された.また遺伝子組換え酵素CreとR8の融合タンパク質(Cre-R8)の細胞への取り込み様式を,各種エンドサイトーシス阻害剤の存在下でのloxP部位における遺伝子組換え効率を指標に検討したところ,やはりクラスリン依存性エンドサイトーシスにより取り込まれることが示唆された.この結果は,R8を用いた細胞内送達において,シンデカン-4を受容体とするエンドサイトーシス系が,細胞外から添加したタンパク質の細胞内活性を導く系であることを示す結果である.これはまたアルギニンペプチドをはじめとするCPPにおいて細胞内活性の発現につながるエンドサイトーシス系とその受容体を同定した初めての例といえる.
アルギニンペプチドは,投与条件に応じ,さまざまな経路で細胞内に移行でき,また,ある経路が阻害されると別の経路を使って細胞内に移行するようになるなど,通常みられる受容体–リガンド相互作用とはかなり違った様式で細胞内に移行する.この際,特にペプチドの濃度が経路決定に重要な働きをする.このようにさまざまな経路を条件に応じて活用して細胞内に移行することができる点が,おそらく,アルギニンペプチドが効率的なCPPとして頻用される理由の一つであると同時に,アルギニンペプチドの細胞内移行機序の解明を困難にする理由の一つと考えられる.今後さらなる検討により,取り込み受容体や取り込み機序に関しての知見が蓄積されることによって,細胞内への物質取り込みに関する新た知見や概念が得られることを期待する.
上述のように,アルギニンペプチドは条件を選ぶことで細胞膜(形質膜)を透過して直接カーゴを細胞内(サイトゾル)に送達することが可能である.このためには,前述のように,アルギニンペプチドが細胞表面に比較的高い濃度で集積することや,カーゴ分子の大きさがあまり大きくないことが必要である.アルギニンペプチドのような正電荷に富む分子が疎水性の膜環境を通過しようとする際には,アルギニンのグアニジノ基が水分子や水中の溶質分子と形成する水素結合やイオン対を解消することが必要となる.これは,エネルギー的に不利な状況であり,高いエネルギー障壁を越える必要が出てくる.しかし,前述のRothbardらの例にもあるように,疎水性の対イオン分子が膜内に存在すると仮定すると,アルギニンペプチドはこの分子と水素結合やイオン対を形成することができ,エネルギー障壁を下げることができる.ジュネーブ大学のSakai, Matileらは独自の観点からこのような対イオンを用いたカチオン性分子の膜内移行について研究を進めており,その結果,芳香環とカルボキシ基,あるいはリン酸基,硫酸基等グアニジノ基と2本の水素結合を形成可能なアニオン基を併せ持つ一連の化合物群存在下にポリアルギニンが脂質二重膜を透過できることを見いだした48).筆者らは同グループとの共同研究により,ピレンブチレートなどの複数の化合物がアルギニンペプチドの細胞内移行を大きく促進することを見いだした.最終濃度50 µM程度となるようにピレンブチレートを細胞に加えた後にR8などのアルギニンペプチドを添加すると,数分以内に,ほぼ100%の細胞にR8の移行が観察された24).またペプチドや緑色蛍光タンパク質GFP等の小型タンパク質とR8の融合タンパク質も同様の効率で細胞内に導入できることがわかった24, 48–51).また,通常遺伝子導入による細胞内タンパク質発現が困難な神経系の初代培養細胞(マウス海馬由来初代神経細胞)にも非常に高い効率でR8とEGFPの融合タンパク質が導入された.血清存在下ではピレンブチレートの血清中のタンパク質への吸着によりこの促進効果が大幅に減弱するので,細胞レベルでの実験に用途は限定されるものの,遺伝子導入と相補的な方法として,細胞生物学やケミカルバイオロジー研究に有用な手段といえる.この方法を用いて,安定同位体ラベルしたタンパク質を細胞内に導入することで,哺乳動物の生細胞中でのタンパク質のNMRも測定された52).
筆者らのグループでは,ペプチドを用いた新しい細胞操作技術の樹立を視野に,細胞膜に形態変化を誘起するペプチドの創出に取り組んでいる.この過程で,エプシン-1のN末端由来の正曲率誘導ペプチドEpN18存在下に,アルギニンペプチドの形質膜の直接透過が亢進されることを見いだした53).エプシン-1はクラスリン依存性エンドサイトーシスにおける細胞内取り込みの初期段階の細胞表面におけるクラスリン被覆ピット(陥入口)形成に関わるタンパク質である54).ホスファチジルイノシトール4,5-ビスリン酸[PI(4,5)P2]にリクルートされて膜に近づくとN末端へリックスを膜内に挿入する.これにより,膜の細胞質側が押し広げられ,細胞表面から見て内側に窪んだ膜構造が形成される.EpN18による直接膜透過機序の詳細に関しては現在検討中であるが,膜が湾曲すると,膜脂質のアシル鎖がより膜表面に露出した膜構造の欠陥状態が増加し,これが膜構造を脆弱にし,アルギニンペプチドの透過を促進する可能性も考えられる.
以上,エンドサイトーシスと直接透過の両面からアルギニンペプチドの膜透過様式について概説した.たとえばクラスリン依存性エンドサイトーシスに基づく受容体依存性エンドサイトーシスでは,多くの場合,リガンドは特定の受容体との特異的な相互作用を介して細胞内に取り込まれるため,他の取り込み経路を考慮する必要がないのが一般的である.これに対して,アルギニンペプチドの膜透過は,相互作用しうる膜表面分子が複数(おそらくは非常に多く)あり,細胞表面に付着したアルギニンペプチドの量やカーゴの物性に応じて主な取り込み経路が変わりうるという点で特徴的である.特定の取り込み経路が阻害剤などによって遮断されると,別の経路で取り込みが開始されるという点でも複雑である.これまでリガンド–受容体相互作用として,解離定数がnMから数十nM程度の比較的強い相互作用を示すものが主に研究されてきたように思われるが,アルギニンペプチドと細胞表面の相互作用分子との相互作用は,おそらく従来は非特異的吸着として扱われてきたもの(解離定数としてサブµMあるいはそれ以上?)であると考えられる.このような複数の弱い相互作用が相加的あるいは相乗的に働くことで細胞内への取り込みが行われている可能性も高いのではないかとも考えられる.アルギニンペプチドはカチオン性のアルギニンが高頻度で配置された特別なペプチドともいえるが,これをプローブとして細胞内物質取り込みのさまざまな局面が明らかとなってきているように思える.細胞内への物質取り込みや薬物送達といった周辺分野への波及効果も含めて,今後さらに研究を進めてゆきたいと考えている.
1) Kurrikoff, K., Gestin, M., & Langel, Ü. (2016) Expert Opin. Drug Deliv., 13, 373–387.
2) Boisguérin, P., Deshayes, S., Gait, M.J., O’Donovan, L., Godfrey, C., Betts, C.A., Wood, M.J., & Lebleu, B. (2015) Adv. Drug Deliv. Rev., 87, 52–67.
3) Lönn, P. & Dowdy, S.F. (2015) Expert Opin. Drug Deliv., 12, 1627–1636.
4) Stanzl, E.G., Trantow, B.M., Vargas, J.R., & Wender, P.A. (2013) Acc. Chem. Res., 46, 2944–2954.
5) Takeuchi, T. & Futaki, S. (2016) Chem. Pharm. Bull. (Tokyo), 64, 1431–1437.
6) Nakase, I., Takeuchi, T., Tanaka, G., & Futaki, S. (2008) Adv. Drug Deliv. Rev., 60, 598–607.
7) Futaki, S. (2005) Adv. Drug Deliv. Rev., 57, 547–558.
8) Vivès, E., Brodin, P., & Lebleu, B. (1997) J. Biol. Chem., 272, 16010–16017.
9) Futaki, S., Suzuki, T., Ohashi, W., Yagami, T., Tanaka, S., Ueda, K., & Sugiura, Y. (2001) J. Biol. Chem., 276, 5836–5840.
10) Wender, P.A., Mitchell, D.J., Pattabiraman, K., Pelkey, E.T., Steinman, L., & Rothbard, J.B. (2000) Proc. Natl. Acad. Sci. USA, 97, 13003–13008.
11) Dupont, E., Prochiantz, A., & Joliot, A. (2011) Methods Mol. Biol., 683, 21–29.
12) Nekhotiaeva, N., Elmquist, A., Rajarao, G.K., Hällbrink, M., Langel, Ü., & Good, L. (2004) FASEB J., 18, 394–396.
13) El-Sayed, A., Khalil, I.A., Kogure, K., Futaki, S., & Harashima, H. (2008) J. Biol. Chem., 283, 23450–23461.
14) Rothbard, J.B., Jessop, T.C., Lewis, R.S., Murray, B.A., & Wender, P.A. (2004) J. Am. Chem. Soc., 126, 9506–9507.
15) Hirose, H., Takeuchi, T., Osakada, H., Pujals, S., Katayama, S., Nakase, I., Kobayashi, S., Haraguchi, T., & Futaki, S. (2012) Mol. Ther., 20, 984–993.
16) Nakase, I., Tadokoro, A., Kawabata, N., Takeuchi, T., Katoh, H., Hiramoto, K., Negishi, M., Nomizu, M., Sugiura, Y., & Futaki, S. (2007) Biochemistry, 46, 492–501.
17) Suzuki, T., Futaki, S., Niwa, M., Tanaka, S., Ueda, K., & Sugiura, Y. (2002) J. Biol. Chem., 277, 2437–2443.
18) Nakase, I., Hirose, H., Tanaka, G., Tadokoro, A., Kobayashi, S., Takeuchi, T., & Futaki, S. (2009) Mol. Ther., 17, 1868–1876.
19) Gonçalves, E., Kitas, E., & Seelig, J. (2005) Biochemistry, 44, 2692–2702.
20) Ziegler, A. & Seelig, J. (2011) Biochemistry, 50, 4650–4664.
21) Tünnemann, G., Martin, R.M., Haupt, S., Patsch, C., Edenhofer, F., & Cardoso, M.C. (2006) FASEB J., 20, 1775–1784.
22) Kosuge, M., Takeuchi, T., Nakase, I., Jones, A.T., & Futaki, S. (2008) Bioconjug. Chem., 19, 656–664.
23) Takayama, K., Hirose, H., Tanaka, G., Pujals, S., Katayama, S., Nakase, I., & Futaki, S. (2012) Mol. Pharm., 9, 1222–1230.
24) Takeuchi, T., Kosuge, M., Tadokoro, A., Sugiura, Y., Nishi, M., Kawata, M., Sakai, N., Matile, S., & Futaki, S. (2006) ACS Chem. Biol., 1, 299–303.
25) Brooks, H., Lebleu, B., & Vivès, E. (2005) Adv. Drug Deliv. Rev., 57, 559–577.
26) Richard, J.P., Melikov, K., Vives, E., Ramos, C., Verbeure, B., Gait, M.J., Chernomordik, L.V., & Lebleu, B. (2003) J. Biol. Chem., 278, 585–590.
27) Nakase, I., Niwa, M., Takeuchi, T., Sonomura, K., Kawabata, N., Koike, Y., Takehashi, M., Tanaka, S., Ueda, K., Simpson, J.C., Jones, A.T., Sugiura, Y., & Futaki, S. (2004) Mol. Ther., 10, 1011–1022.
28) Wadia, J.S., Stan, R.V., & Dowdy, S.F. (2004) Nat. Med., 10, 310–315.
29) Meier, O. & Greber, U.F. (2003) J. Gene Med., 5, 451–462.
30) Brock, R. (2014) Bioconjug. Chem., 25, 863–868.
31) Kawaguchi, Y., Takeuchi, T., Kuwata, K., Chiba, J., Hatanaka, Y., Nakase, I., & Futaki, S. (2016) Bioconjug. Chem., 27, 1119–1130.
32) Conner, S.D. & Schmid, S.L. (2003) Nature, 422, 37–44.
33) Kerr, M.C. & Teasdale, R.D. (2009) Traffic, 10, 364–371.
34) Bauer, A., Subramanian, N., Villinger, C., Frascaroli, G., Mertens, T., & Walther, P. (2016) Histochem. Cell Biol., 145, 617–627.
35) Mercer, J. & Helenius, A. (2009) Nat. Cell Biol., 11, 510–520.
36) Nakase, I., Osaki, K., Tanaka, G., Utani, A., & Futaki, S. (2014) Biochem. Biophys. Res. Commun., 446, 857–862.
37) Pellegrini, L. (2001) Curr. Opin. Struct. Biol., 11, 629–634.
38) Mitola, S., Sozzani, S., Luini, W., Primo, L., Borsatti, A., Weich, H., & Bussolino, F. (1997) Blood, 90, 1365–1372.
39) Rubio Demirovic, A., Canadi, J., Weiglhofer, W., Scheidegger, P., Jaussi, R., & Kurt, B.H. (2003) Biol. Chem., 384, 1435–1441.
40) Xiao, H., Neuveut, C., Tiffany, H.L., Benkirane, M., Rich, E.A., Murphy, P.M., & Jeang, K.T. (2000) Proc. Natl. Acad. Sci. USA, 97, 11466–11471.
41) Tanaka, G., Nakase, I., Fukuda, Y., Masuda, R., Oishi, S., Shimura, K., Kawaguchi, Y., Takatani-Nakase, T., Langel, Ü., Gräslund, A., Okawa, K., Matsuoka, M., Fujii, N., Hatanaka, Y., & Futaki, S. (2012) Chem. Biol., 19, 1437–1446.
42) Nakashima, H., Hashimoto, M., Sadakane, Y., Tomohiro, T., & Hatanaka, Y. (2006) J. Am. Chem. Soc., 128, 15092–15093.
43) Rey, M., Valenzuela-Fernández, A., Urzainqui, A., Yáñez-Mó, M., Pérez-Martínez, M., Penela, P., Mayor, F. Jr., & Sánchez-Madrid, F. (2007) J. Cell Sci., 120, 1126–1133.
44) Wu, Y. & Yoder, A. (2009) PLoS Pathog., 5, e1000520.
45) Kawaguchi, Y., Tanaka, G., Nakase, I., Imanishi, M., Chiba, J., Hatanaka, Y., & Futaki, S. (2013) Bioorg. Med. Chem. Lett., 23, 3738–3740.
46) Verhelst, S.H., Fonović, M., & Bogyo, M. (2007) Angew. Chem. Int. Ed., 46, 1284–1286.
47) Yang, Y.Y., Grammel, M., Raghavan, A.S., Charron, G., & Hang, H.C. (2010) Chem. Biol., 17, 1212–1222.
48) Sakai, N. & Matile, S. (2003) J. Am. Chem. Soc., 125, 14348–14356.
49) Nishihara, M., Perret, F., Takeuchi, T., Futaki, S., Lazar, A.N., Coleman, A.W., Sakai, N., & Matile, S. (2005) Org. Biomol. Chem., 3, 1659–1669.
50) Perret, F., Nishihara, M., Takeuchi, T., Futaki, S., Lazar, A.N., Coleman, A.W., Sakai, N., & Matile, S. (2005) J. Am. Chem. Soc., 127, 1114–1115.
51) Sakai, N., Takeuchi, T., Futaki, S., & Matile, S. (2005) ChemBioChem, 6, 114–122.
52) Inomata, K., Ohno, A., Tochio, H., Isogai, S., Tenno, T., Nakase, I., Takeuchi, T., Futaki, S., Ito, Y., Hiroaki, H., & Shirakawa, M. (2009) Nature, 458, 106–109.
53) Pujals, S., Miyamae, H., Afonin, S., Murayama, T., Hirose, H., Nakase, I., Taniuchi, K., Umeda, M., Sakamoto, K., Ulrich, A.S., & Futaki, S. (2013) ACS Chem. Biol., 8, 1894–1899.
54) Itoh, T. & De Camilli, P. (2006) Biochim. Biophys. Acta, 1761, 897–912.

京都大学化学研究所教授.薬学博士.
1983年京都大学薬学部卒.87年同大学院薬学研究科博士後期課程中退,徳島大学薬学部助手.93年同助教授,97年京都大学化学研究所助教授,2005年より現職.この間,89~91年米国ロックフェラー大学博士研究員.
研究テーマと抱負膜と相互作用するペプチドの設計.ペプチドをツールとして細胞を操る新しい手法を開発したいと思っています.
ウェブサイトhttp://www.scl.kyoto-u.ac.jp/~bfdc/index.html
趣味知らない土地を歩くこと.

This page was created on 2016-12-27T09:08:26.531+09:00
This page was last modified on 2017-02-17T18:15:58.355+09:00
このサイトは(株)国際文献社によって運用されています。