発達障害・精神神経疾患はさまざまな社会問題の一因となるため,近年これら疾患の発症メカニズムの解明や有効な治療法および診断・予防法の開発に対する社会的な需要がよりいっそう高まっている.自閉症スペクトラム障害(autism spectrum disorder:ASD)は,社会性・コミュニケーション能力の障害,繰り返し行動や偏った興味などにより特徴づけられる複雑な神経発達障害である.ASDは異なる遺伝的・環境的要因の組合わせで発症すると考えられており,その病態はきわめて複雑である1).最新の研究により,アメリカにおけるASDの発症頻度は8歳以下で68人に1人と報告されている2).このように高頻度な発症率にも関わらず,依然としてASDの発症病態は大部分が不明瞭であるが,その発症に関与する特定遺伝子機能欠損の研究は,ASD病態の共通メカニズムの解明につながることが期待されている.
ASDの一つであるレット症候群(Rett syndrome:RTT)は,1966年にウィーンの小児神経科医Andreas Rettにより初めて症例が報告された3).その後,1980年代に多くの症例が報告されるようになり4),レット症候群は主に女児に発症する神経発達障害として世界的に認知されるようになった.一方で,1980年代から,DNAメチル化,ヒストン修飾などのエピジェネティックな修飾が遺伝子発現を制御することが次々と明らかにされ,さまざまな生命現象におけるエピジェネティクスの重要性が確立されてきた.methyl-CpG binding protein 2(MeCP2)は,1992年にBirdらによって哺乳類のメチル化されたDNAに強い親和性を持って結合するメチル化DNA結合タンパク質として初めて同定され5),その後の研究により,MeCP2がメチル化された標的遺伝子に結合し,その発現を抑制する活性を持つことが報告された6).1999年,Hudaらのグループにより多くのRTT患者がMeCP2遺伝子に変異を有することが示され,MeCP2がRTTの原因遺伝子であることが明らかとなった7).この報告以来,MeCP2はエピジェネティクスと脳機能・神経疾患を結びつける分子として非常に注目され,世界中で精力的に研究されるようになった.その後,MeCP2遺伝子の変異はRTT患者だけでなく,幅広い精神疾患患者にも認められることやMeCP2遺伝子の重複も発達障害を引き起こすことなどが明らかになり,病態発症に重要なMeCP2の分子機能を解明することは広範囲の精神・発達障害を理解することに寄与すると考えられるようになった8).本稿ではまず前半に,エピジェネティクスについて,筆者らのこれまでの研究成果を交えて概説する.中盤においてレット症候群とMeCP2の機能についてこれまでに明らかになっている知見を紹介する.後半では我々の最新の研究により明らかとなったMeCP2の新しい機能「microRNA(miRNA)プロセシング促進作用」とそのレット症候群病態への寄与について概説したい.
エピジェネティクスとは,DNA塩基配列上の変化を伴わずに遺伝情報の発現を調節する機構のことである.この調節機構にはクロマチン構造因子であるDNAのメチル化修飾,ヒストンのアセチル化・メチル化等のタンパク質の翻訳後修飾,クロマチン再構築酵素群によるATP依存的なクロマチン構造変換,さらにnon-coding RNA(ncRNA)による転写や翻訳後修飾などが含まれる.
ヒトを含めた哺乳類のゲノムDNAでは5′-CpG-3′の2塩基配列のシトシンが5位炭素原子においてメチル化修飾を受けることが知られている.遺伝子発現におけるDNAメチル化の寄与として,遺伝子プロモーター中のシトシンがメチル化されると遺伝子発現が抑制されることが知られている.このメカニズムは大きく二つに分けられ,一つは,転写因子の結合配列中のCpG配列がメチル化された結果,転写因子のプロモーターへの結合が妨げられる機構,もう一つはMeCP2などに代表されるメチル化DNA結合タンパク質がメチル化されたDNA配列に結合し,ヒストン脱アセチル化酵素(histone deacetylase:HDAC)などのリプレッサー因子をリクルートすることで遺伝子発現を抑制する機構である.
DNAメチル化に加えて,ヒストン修飾もエピジェネティクスの代表的な制御基盤である.クロマチン構造の最小単位はヌクレオソームで,ヒストンH2A, H2B, H3, H4それぞれ2分子ずつからなる八量体の構造をとっており,DNAがその周りを左巻きに巻いている.このヒストンのN末端領域は立体構造に乏しく,リン酸化,アセチル化,メチル化,ユビキチン化,ADPリボシル化,グリコシル化など多様なアミノ酸修飾を受け,クロマチン構造の変化に関与している.さらに,ATP依存的にヌクレオソームの構造変換を引き起こすSWI/SNF(mating type switching/sucrose nonfermenting)複合体などのクロマチンリモデリング酵素群が存在し,それらがヒストンタンパク質をスライドさせたり,ヒストンとDNAの凝集を防いだりすることにより,遺伝子の転写調節を行っている.
また,近年ではmiRNAを含めたncRNAによる遺伝子発現制御が大きな注目を集めている.細胞内には,タンパク質へと翻訳されないncRNAが大量に存在する.ncRNA群はその長さや機能などからさまざまなカテゴリーに分類される.これらのncRNAの中でも低分子RNAの代表の一つであるmiRNAは,転写後の遺伝子発現制御において重要な役割を果たしている.miRNAは21~25塩基ほどの短鎖ncRNAであり,内在性RNAサイレンシング機構を担う代表的な低分子RNA群である.現在,miRNAは多彩な生命現象,および種々の疾患の分子病態に寄与していることが明らかにされており,精力的に研究が行われている.
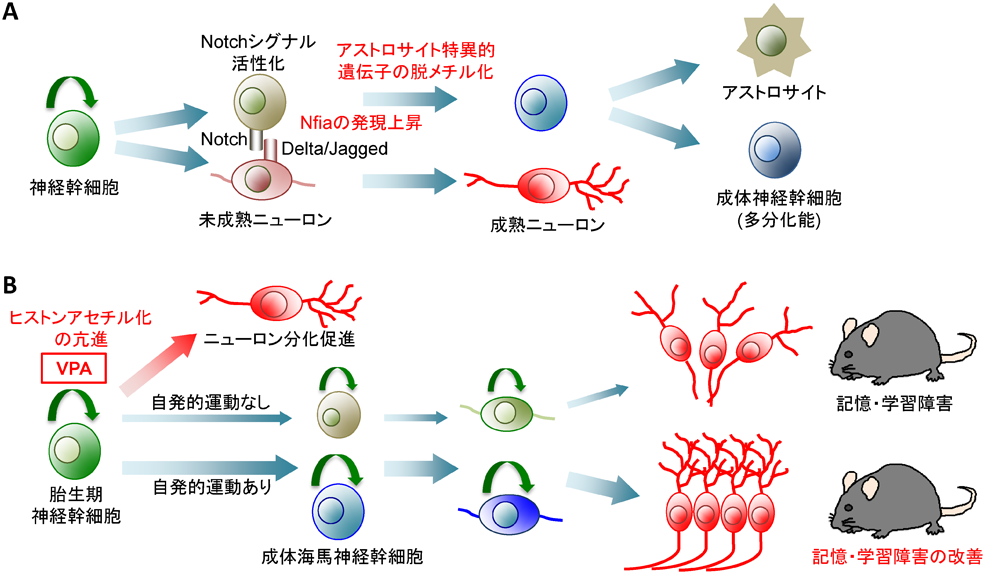
筆者らはこれまで中枢神経系の主要細胞種を産生する神経幹細胞の運命決定におけるエピジェネティックな制御基盤の研究を行ってきた.マウス中枢神経の発生過程において,神経幹細胞は胎生中期には主にニューロンへと分化し,胎生後期以降はニューロン分化よりもアストロサイトへの分化が優位になることが知られている9).筆者らは,この神経幹細胞の発生段階依存的なアストロサイト分化に,アストロサイト特異的遺伝子の転写調節領域のDNAメチル化が関与していることを明らかにしてきた9).胎生中期の神経幹細胞では,アストロサイト特異的発現遺伝子glial fibrillary acidic protein(Gfap)の転写調節領域が高頻度にメチル化されているが,胎生後期になるとこの部位の脱メチル化が生じ,転写因子signal transducer and activator of transcription 3(STAT3)のGfap転写調節領域上の認識配列への結合が可能となる.そのため,サイトカインなどによるSTAT3の活性化に応じてGfapを発現するアストロサイトへと分化が可能となる.同様の現象は,他のアストロサイト特異的遺伝子S100βのプロモーター領域においても観察される.さらに我々は,この脱メチル化のメカニズムとして,神経幹細胞から先に産生されるニューロンがNotchリガンドであるDelta like 1及びJagged 1を発現しており,神経幹細胞のNotchシグナルを活性化すること,活性化されたNotchシグナルがnuclear factor i-A(Nfia)タンパク質の発現を誘導し,アストロサイト特異的遺伝子プロモーターの脱メチル化が引き起こされることを明らかにした(図1A)10).これらの知見は,DNAメチル化が発生段階依存的な神経幹細胞の多分化能獲得において重要な鍵となっていることを示している.
ヒストンアセチル化酵素(histone acetyltransferase:HAT)は,ヒストン尾部にアセチル基を付加することで,ヒストンの陽電荷を減少させ,陰に荷電しているDNAとの相互作用を弱めることでクロマチン構造を脱凝縮した状態にし,転写の活性化を促進する.逆にヒストン脱アセチル化酵素(HDAC)はヒストン尾部のアセチル基を取り除く.これによりDNAとヒストンの親和性が増強することでクロマチン構造は閉じた状態になり,転写が抑制された状態になる.抗てんかん薬であるバルプロ酸(valproic acid:VPA)は,HDAC阻害剤としての活性を有していることが知られている11).筆者らは,成体ラット海馬由来の培養神経幹細胞をVPAで処理すると,ニューロン分化を促進する転写因子NeuroDの発現が亢進し,高効率なニューロン分化が誘導されることを報告した12).生体内においても,ラットへのVPA投与により,海馬に存在する神経幹細胞の増殖が抑制され,ニューロン分化が亢進された12).また,我々は最近,VPAを妊娠マウスに投与した場合,出生・成長した子どもの脳ではニューロン産生能が低下してしまうため,学習・記憶が障害されること,この学習・記憶能の低下は自発的運動によって改善されることを報告した(図1B)13).
以上のように,これまでに筆者らはエピジェネティックな制御機構が神経系細胞の分化制御にきわめて重要な役割を果たしていることを明らかにしてきた.本稿後半においては,我々が新規に発見したMeCP2によるmiRNA生合成を介した神経機能制御について紹介するが,まずは次節でこのMeCP2が原因因子となっているレット症候群とこれまでに明らかになっていたMeCP2の作用について解説したい.
3. レット症候群(RTT)とその原因因子MeCP2の作用
1)RTTの臨床症状
RTTは,主に女児にみられる進行性の重篤な精神・神経発達障害である.RTTはX連鎖優性遺伝病で10,000人から15,000人に1人の割合で発症する.RTT患者は,生後6か月から18か月程度までは正常に発達するが,その後,それまでに獲得された言語能力や運動機能の退行がみられるようになる.RTT患者は短寿命ではあるものの,大半は成人まで生存し,自閉傾向やてんかん,手もみ運動に代表される常同行動や精神遅滞,小頭症などの種々の神経学的症状を示す.神経系以外の症状としては,全身の成長遅延や骨形成不全などの特徴がみられる3, 4).
2)RTTの遺伝学的背景
RTTは主に女性にみられることからX染色体連鎖型の優性遺伝であることが想定されていた.RTT症例の99%以上が弧発性であったことから,連鎖解析による染色体上の病態責任領域の位置づけは非常に困難であったが,希少な家族性症例の情報を使った排他的マッピングによりXq28が候補領域として同定され,その後のRTT患者における候補遺伝子のスクリーニングによりMeCP2遺伝子の変異が特定された7).
3)MeCP2遺伝子の構造と変異
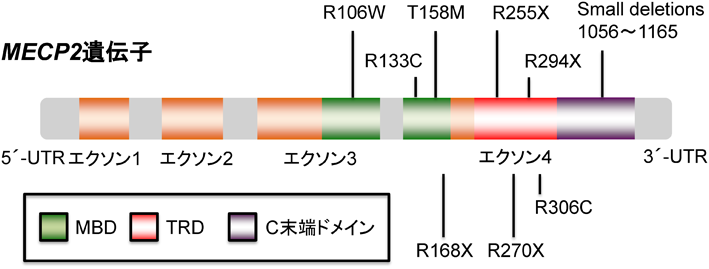
MeCP2遺伝子(MECP2)は,メチル化DNA結合ドメイン(MBD),転写抑制ドメイン(TRD),C末端ドメインの三つの機能ドメインを有している.MeCP2遺伝子の変異は,95%以上の古典的RTT症例に認められる.古典的RTTの臨床診断基準として,以下の七つの項目が示されている.①出生前および周産期は一見異常がみられない,②出生時の頭囲は正常,③6か月までは発達に異常がみられない,④3~48か月の間に頭囲の発育に停滞がみられる,⑤5~30か月の間に,一度獲得された合目的な手の動きが消失し,引き続き手の同一運動の出現がみられる,⑥重度の精神遅滞とともに,言語の表出,認知の重度の障害がみれれる,⑦12~48か月の間に歩行失調,体幹失調の進行がみられる.MeCP2遺伝子変異のタイプは,T158MやR306Cを代表とするミスセンス,R168XやR255X,R270Xなどのナンセンスやフレームシフト型など,300以上のヌクレオチド置換が報告されている(図2)14).重要なことに,MeCP2遺伝子の変異はRTTだけでなく,知能障害,自閉症,双極性障害,統合失調症などの幅広い精神疾患患者にも見いだされている.また,MeCP2遺伝子の重複も神経発達障害を引き起こすことが明らかになっている.このことは,MeCP2の適切な量が正常な脳機能に重要であるということを示唆している8).
4)RTT動物モデル
RTT病態を理解するために,これまでにさまざまなRTTモデルマウスが作製されてきた.最初に,BirdらとJaenishらの二つの独立したグループによって,エクソン3あるいはエクソン3と4を欠失したMeCP2ノックアウト(KO;MeCP2−/y)マウスが作製された15, 16).これらのRTTモデルマウスはRTT患者の症状を模倣した表現型を示す.MeCP2KOマウスは3~6週齢までは一見正常に発達するが,その後,低活動性や振戦,歩行失調,イレギュラーな呼吸などの神経系症状を示すようになり8~10週齢までに致死となる.MeCP2KOマウスの脳は,RTT患者脳と同様に,全体的な脳体積が小さく,個々のニューロンの細胞体も小さい.また,海馬などを含めた脳のさまざまな領域においてニューロンが密に配置されるという組織学的な特徴を示すが,脳全体の構造的異常は確認されていない.雌のMeCP2+/−ヘテロKOマウスはRTT様の行動異常を示すが,すべての細胞でMeCP2を欠く雄のMeCP2−/yマウスよりも遅い時期に表現型が観察される.さらにNestin-Creドライバーを用いて作製された中枢神経系特異的MeCP2欠損マウスは,全身性のMeCP2KOマウスと類似した表現型がみられることから,脳・神経系におけるMeCP2の欠損は,RTTの病態を引き起こすのに十分であることが証明された16).また,Calcium-calmodulin-dependent kinase II(CamKII)-Creドライバーを用いて作製された最終分裂後のニューロンに特異的なMeCP2欠損マウスでも,時期は遅延するものの類似のRTT表現型がみられることから,成熟ニューロン機能におけるMeCP2の重要性が確認されている15, 17).重要なことに,MeCP2KOマウスにおいて神経系細胞およびニューロンにMeCP2を再発現させると,上述のMeCP2KOマウスのRTT表現型が改善されることが示された18, 19).これは,進行性の発達障害を治療することが可能であることを示唆する非常に重要な知見である.上述のモデルマウスに加えて,実際のRTT患者でみられるMeCP2変異体を発現するノックインマウスが次々と作製されている.RTT患者で高頻度にみられるC末端欠失型ナンセンス変異体を模倣したMeCP2308/yマウスは,運動機能障害,低活動,社会行動異常,不安様行動などのRTT患者でみられる多くの表現型を示す20–22).点変異型ミスセンス変異体を発現するMeCP2T158M/yマウス23)や欠失変異型ナンセンス変異体MeCP2R168X/yマウス24)も作製されており,部分的なRTT表現型を示すことが報告されている.また,MeCP2遺伝子を重複したトランスジェニックマウスも作製されており,MeCP2の異常発現亢進によって引き起こされるヒトの神経発達障害の表現型を模倣することが報告されている25).また最近では,中国のグループによりMeCP2遺伝子の重複を模倣した霊長類モデルが作製されており26),今後は霊長類を用いた解析により,MeCP2機能異常に起因する病態のさらなる理解が期待される.
5)MeCP2機能不全による表現型
a. 神経病理学的異常
RTTは重篤な神経機能障害を呈するが,意外にも中枢神経系における主な病理学的所見としては,脳体積の減少とニューロンの細胞体サイズの減少がみられる程度であり,全体的な脳構造に異常は見いだされていない.検死解剖による解析からRTT患者の脳は,健常者と比較し12~13%の重量と体積の減少を示すことが報告されている27).RTT脳では炎症や変性,グリオーシスや神経細胞移動などの異常は観察されないことから,RTTは神経変性疾患ではなく神経発達障害であると考えられている28, 29).また,大脳皮質第IIIおよび第V層の錐体ニューロンにおける樹状突起の枝分かれの減少27),前頭皮質における樹状突起スパインの形態が短く,低密度に散在していることが報告されている30).さらに,皮質,視床,扁桃体,海馬におけるニューロン細胞体サイズの減少がみられることに加え,ニューロンの高密度にパックした局在が観察されている.これらのRTT患者における神経病理学的所見は,MeCP2KOマウスにおいてもその多くが再現されている15, 16).
b. 神経生理学的異常
RTT患者における神経生理学的研究から,RTT脳の皮質において異常な体性感覚誘発電位や脳波などの興奮性の変質が報告されている31).RTTマウスモデルを用いた研究から,MeCP2-/yマウスやMeCP2308/yマウスの皮質スライスでは,長期増強(long-term potentiation:LTP)の減少がみられること32, 33),MeCP2-/yマウスの皮質においては,興奮性シナプス伝達の減少と抑制性シナプス伝達の上昇が示されている34).また,MeCP2-/yマウス由来の培養海馬ニューロンは自発的な興奮性シナプス伝達の頻度の減少と興奮性シナプス形成(密度)の減少を示すことが報告されている35, 36).これらの報告から,MeCP2は主に興奮性シナプス伝達の制御に重要な役割を担っていることが示唆される.抑制性シナプス伝達の制御におけるMeCP2の役割については今後の研究が待たれる.
6)MeCP2遺伝子の発現と分子機能
a. MeCP2遺伝子の発現様式
MeCP2遺伝子は四つのエクソンを有し,エクソン2の選択的スプライシングのため,二つの異なるタンパク質アイソフォームが産生される(図3).MeCP2の二つのスプライシングバリアントは,N末端部分のみが異なる遺伝子構造をとる.MECP2αによりコードされるMeCP2-e1アイソフォームは,生体内でより豊富に存在し,エクソン1の24アミノ酸を含み,エクソン2の9アミノ酸を欠いている.一方,MECP2βによりコードされるMeCP2-e2は,エクソン2より転写される37, 38)(図3).さらにMeCP2遺伝子は,複数のポリ(A)付加部位を含むヒトとマウス間で高度に保存された3′-UTRを有している.MeCP2のmRNA発現解析により,最も長い転写産物が脳において豊富に存在することが明らかにされた.この転写産物は胎生期に発現が高く,生後にいったん発現が減少するが,その後生体において再度発現が上昇するという興味深い発現パターンを示す39).MeCP2タンパク質の発現量は,胎生期には低く,生後のニューロンの成熟に伴って高くなることが示されている40).
b. MeCP2の分子機能
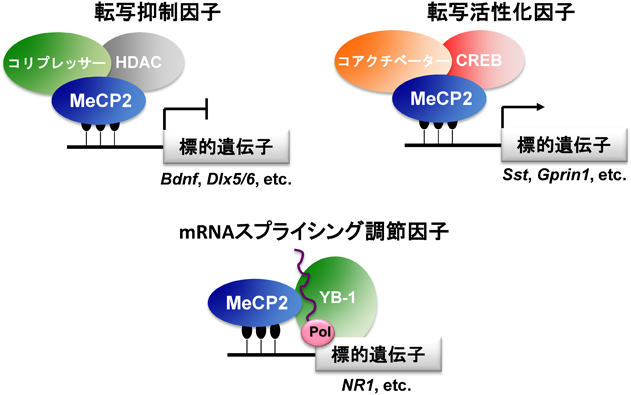
MeCP2タンパク質におけるMBDはメチル化されたCpGヌクレオチドに特異的に結合し,このMeCP2のCpGヌクレオチドに対する結合特異性は近接するA/Tリッチな配列に依存することが報告されている41).TRDはコリプレッサーやクロマチンリモデリング因子をリクルートすることにより転写抑制活性に寄与する.C末端ドメインはnaked DNAやヌクレオソームへのMeCP2の結合を促進することが示されているが,詳細な機能的役割は明らかになっていない42).しかしながら,多くのRTT患者においてC末端領域の欠失がみられること,MeCP2のC末端領域を欠失したモデルマウスは種々のRTT表現型を再現することからRTT病態におけるMeCP2の正常な機能に重要なことは明らかである.
前述のように,当初からMeCP2はメチル化された遺伝子プロモーターに結合し標的遺伝子の発現を抑制する転写抑制因子として作用すると考えられてきた.この転写抑制には,ヒストン脱アセチル化酵素(HDAC)やmSin3AなどのコリプレッサーやSWI/SNFクロマチンリモデリング複合体の構成因子Brahma, DNAメチル基転移酵素Suv39H1などとの相互作用により媒介されることが示されている43–45).しかしながら,RTT病態におけるこれらのタンパク質との相互作用の機能的意義は明らかになっていない.MeCP2はこれまでの研究により転写抑制因子として作用するだけでなく,さまざまな機能を持つことが次々と明らかになっている.MeCP2はクロマチンループ構造の形成を介してクロマチンの凝集を制御することや46),RNA結合タンパク質であるY-box-binding protein 1(YB-1)と相互作用し,mRNAのスプライシングを調節すること47),さらに転写因子cAMP response element binding protein 1(CREB1)と結合し転写活性化因子としても機能すること48)が報告された.このようにMeCP2はさまざまなタンパク質と相互作用することで,非常に多様な機能を持つことが明らかになりつつある(図4).ごく最近では,MeCP2がヒドロキシメチル化シトシン(5hmC)にも結合することが示され,RTT患者でみられるMECP2変異体R133Cでは5hmCとの結合が減弱することから,この機能の病態メカニズムへの関与も示唆されている49).今後の詳細な研究により,RTT病態との関連が明らかになることが期待される.
7)MeCP2下流標的因子
RTTを含めたMeCP2の機能異常に起因する精神・発達障害の病態解明や治療法開発を考える上でMeCP2の下流標的因子を同定することは必須である.MeCP2がメチル化DNA結合タンパク質であり転写抑制因子として考えられていたことから,RTTの疾患病態はMeCP2により本来抑制されるべき標的遺伝子の発現異常亢進が原因と考えられてきた.MeCP2機能異常による標的遺伝子発現異常を調べるために,MeCP2KOマウスの脳組織を用いた転写プロファイリングが行われたが,予想外に,大きな発現変化は検出されず,RTT病態に関連するような異常発現遺伝子の同定には至らなかった.
これまでにMeCP2の転写抑制標的としてbrain-derived neurotrophic factor(BDNF)50)やFXYD domain containing ion transport regulator(FXYD1)51),insulin like growth factor binding protein(IGFBP3)52)遺伝子などが同定されている.BDNFはMeCP2の代表的な下流標的遺伝子であるが,MeCP2KOマウスにおいて発現減少がみられること53)から転写活性化の標的にもなっている可能性があり,MeCP2によってどのように発現が調節されているのか,RTT病態においてどのような役割を果たしているのかは依然として不明瞭である.一方,CREBを介したMeCP2の転写活性の標的としては,somatostatin(Sst)やG protein regulated inducer of neurite outgrowth 1(Gprin1)などが報告されている48).MeCP2のスプライシング制御因子としての標的はglutamate ionotropic receptor NMDA type subunit 1(NR1)などがあげられる47).このように,MeCP2の各種分子機能におけるそれぞれの下流標的が同定されてきているが,これら下流標的因子の発現あるいは機能阻害によりMeCP2KOニューロンの表現型を改善できるような機能的にも信頼できる下流標的は報告されておらず,RTT病態との明確な関連は明らかになっていない.
4. MeCP2によるmiRNAプロセシングを介したmTORシグナル制御
上述してきたように,MeCP2はメチル化DNAに結合し,標的遺伝子の発現を抑制すると報告されていたため,RTTは「メチル化されたMeCP2標的遺伝子の発現異常により発症する」と考えられてきた.しかしこれまでにRTTの表現型と直結する標的遺伝子発現異常は見いだされておらず,MeCP2の機能異常により重篤な神経機能障害が引き起こされる分子メカニズムを説明することができなかった.一方で近年の研究により,発達障害を含めた種々の精神疾患の発症には,mechanistic target of rapamycin(mTOR)シグナルの制御不全が深く関与することが示唆されている54).最近,MeCP2KOマウスおよびヒトMeCP2欠損細胞においても,このmTORシグナルの減弱がみられることが相次いで報告された55, 56).しかし,MeCP2がどのようにmTORシグナルを制御しているのかは明らかにされていなかった.そのため,筆者らは精神・発達障害発症に深く関与するMeCP2の分子作用機序,およびmTORシグナルとの分子相関を明らかにすることで,MeCP2遺伝子の機能異常が原因で引き起こされるRTTの発症メカニズムに迫ろうと考えた57).
1)MeCP2とmiRNAマイクロプロセッサーDrosha複合体の会合
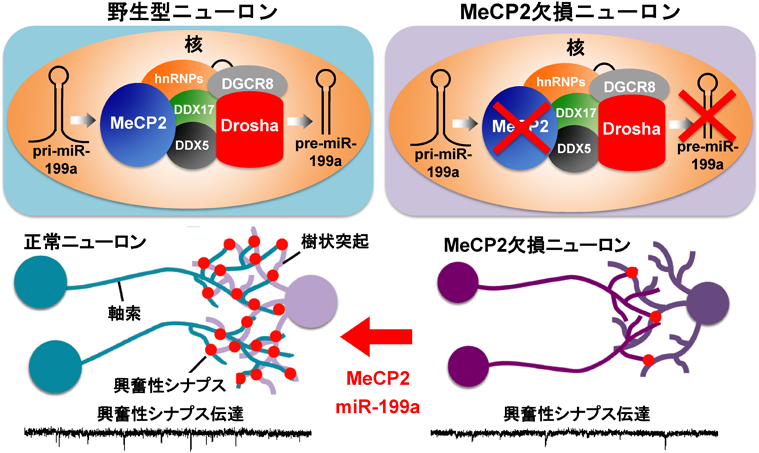
まず筆者らは,MeCP2とmTORシグナルを結びつけるような新規分子機能の同定を目的として,MeCP2相互作用因子の網羅的プロテオミックスクリーニングを実施した.具体的には,中枢神経系の主要細胞種である神経幹細胞,ニューロン,アストロサイト,オリゴデンドロサイトを高純度に培養し,それぞれの細胞種にFLAG-MeCP2を発現するレンチウイルスを感染させ,FLAG抗体を用いた免疫沈降を行った.MeCP2と共沈降したタンパク質を液体クロマトグラフィーハイスループット質量分析により解析した.その結果,これらの中枢神経系主要細胞種における共通のMeCP2相互作用因子として,DDX5/17やhnRNPタンパク質などのmiRNAマイクロプロセッサーであるDrosha複合体の構成因子が同定された.これらのタンパク質は,マウス海馬の抽出液を用いた内在性MeCP2に対する免疫沈降においても検出されたことから,内在性MeCP2ともin vivoにおいて確かに結合することが示唆された.これらのことから,MeCP2がmiRNAのプロセシングにおいて何らかの役割を果たしていることが考えられた.
miRNAは核内においてDNAから長鎖のprimary-miRNAとして転写され,primary-miRNAはDrosha複合体によりヘアピン中間体であるprecursor-miRNAへとプロセシングされる.その後,precursor-miRNAは細胞質へと移行され,Dicer複合体によりmature-miRNAへとさらにプロセシングされる.mature-miRNAはRISCに取り込まれ,標的mRNAの分解や翻訳阻害を誘導する.核内においてprimary-miRNAからprecursor-miRNAへのプロセシングを制御するDrosha複合体は,DGCR8やDEAD-box RNA helicase DDX5/17やhnRNPタンパク質を含めた種々のRNA結合タンパク質により構成されている.これまでの報告により,転写因子Smadやがん抑制因子p53が,DDX5/17を介してDrosha複合体と会合し,細胞外シグナル依存的に特定のmiRNAプロセシングを制御することが報告されている58).筆者らは,MeCP2相互作用因子としてDrosha複合体構成因子であるDDX5/17やhnRNPが同定されたことから,MeCP2がこれらのタンパク質を介してDrosha複合体と会合し,特定miRNAの生合成,すなわち転写後のprimary-miRNAからprecursor-miRNAへのプロセシングを制御するのではないかという仮説を立てた.この仮説を証明するために,ゲル濾過クロマトグラフィーなどを用いた生化学的手法によりMeCP2複合体の精製を行った.イムノブロッティングや質量分析解析により,このMeCP2複合体はDrosha複合体の構成因子を含んでいることが確認された.海馬の抽出液を用いてゲル濾過クロマトグラフィーを行ったところ,内在性のMeCP2とDGCR8, DDX5が同一画分に検出された.これらの結果は,MeCP2がDrosha複合体と会合していることを示唆している.
ごく最近,MeCP2がDGCR8と直接相互作用し,DroshaとDGCR8の会合を阻害することでmiRNAのプロセシングを抑制することが報告された59).しかし,筆者らがMeCP2KOニューロンおよびMeCP2過剰発現ニューロンにおいてDrosha複合体の会合を調べたところ,両者のニューロンにおいてDrosha複合体の会合に変化はみられなかった57).MeCP2のこの作用がRTT病態にどのように関与しているのかは不明である.
2)MeCP2-Drosha複合体の標的miRNA
次に筆者らはMeCP2-Drosha複合体のプロセシング標的miRNAの同定を試みた.野生型とMeCP2KOニューロンおよび神経幹細胞それぞれから抽出したsmall non-coding RNA画分を用いて次世代RNAシークエンス解析を行った.当初の予想どおり,両細胞種において全体的な大きなmature-miRNAの発現変化は検出されなかったが,筆者らは,MeCP2とDrosha構成因子の会合が両細胞種でみられたことから,両細胞種において共通して1.5倍以上発現が減少しているmiRNAに着目した結果,九つのmiRNAを同定した.ここで,MeCP2の下流で作用し,かつmTORシグナルを制御するmiRNAを同定するために機能的スクリーニングを行った.mTORシグナルは細胞体サイズを正に制御することが知られており,MeCP2の過剰発現はmTORシグナルを増大させるためニューロン細胞体サイズを増加させる.そのため,MeCP2標的候補の九つのmiRNAを野生型培養海馬ニューロンにそれぞれ発現させ,細胞体サイズを評価した.そして,九つのmiRNAの中で,唯一miR-199aがMeCP2と同程度に細胞体サイズを増加させることを突き止めた.この結果は,miR-199aがMeCP2の下流で作用することに加え,mTORシグナルを制御することを示唆している.
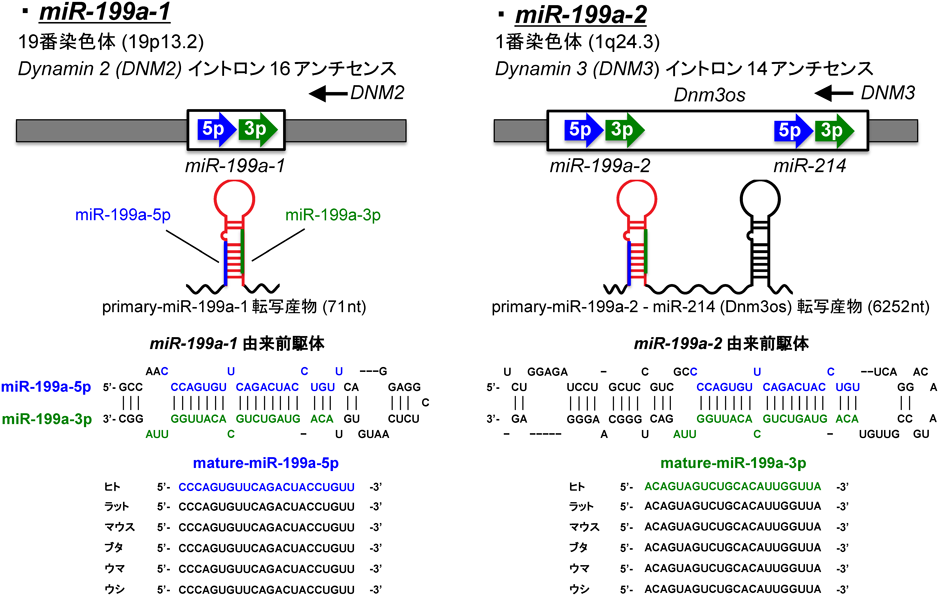
3)miR-199a遺伝子
筆者らがMeCP2の下流標的として同定したmiR-199aは,ゲノム上の二つの遺伝子座miR-199a-1, miR-199a-2にコードされている(図5).miR-199a-1座位からはmiR-199aのみが転写されるが,miR-199a-2座位からはmiR-199aとmiR-214という二つのmiRNAが一つの転写産物として転写される.precursor-miR-199aから,二つの最終産物であるmature-miR-199a-5pと3pが産生される(図5).両者とも多くの哺乳類において高度に保存されているが,神経系におけるmiR-199aの機能はこれまでまったく報告されていなかった.
4)MeCP2によるmiR-199aプロセシング制御
次に筆者らは,MeCP2がmiR-199aを実際に標的とするかどうかを検証するため,まず野生型およびMeCP2KO培養海馬ニューロンにおいてmiR-199aの発現量を定量的リアルタイムPCR(qRT-PCR)により調べた.その結果,MeCP2KOニューロンにおいてmature-miR-199aの有意な発現量の減少が認められ,またMeCP2を過剰発現したニューロンにおいてはmature-miR-199aの発現量亢進がみられた.しかしながらprimary-miR-199aに関しては,mature-formのような顕著な発現量変化は検出されなかった.興味深いことに,RTT患者の死後脳組織においてもmature-miR-199aの有意な発現量の減少が観察された.このことは,miR-199aの制御不全がRTTの病態に関与する可能性を示唆している.また,筆者らはMeCP2抗体を用いたRNA-immunoprecipitation(RIP)アッセイにより,MeCP2とprimary-miR-199aとの細胞内における相互作用を確認した.さらに,in vitro primary-miRNAプロセシングアッセイにより,MeCP2複合体がprimary-miR-199aを特異的にプロセシングすることを明らかにした.これらの結果は,MeCP2複合体がprimary-miR-199aを標的とし,実際にプロセシングを制御できることを示している57).
5)MeCP2下流標的miR-199aの機能
これまでの先行研究により,MeCP2KO培養海馬ニューロンは,細胞体サイズ,興奮性シナプス伝達,興奮性シナプス密度の減少を示すことが明らかにされている.そこで,miR-199aがMeCP2の下流で実際に機能しているのかを検証するために,MeCP2KOニューロンにmiR-199aを発現させ,各種表現型を評価した.その結果,MeCP2KOニューロンにおけるmiR-199aの発現は,減少した細胞体サイズ,興奮性シナプス伝達,シナプス密度を野生型と同程度まで回復させた.さらに,MeCP2を過剰発現するニューロンにおいて内在性miR-199aの機能阻害を行ったところ,MeCP2発現により誘発される細胞体サイズ,興奮性シナプス伝達,シナプス密度の増大が消失された.これらの結果より,MeCP2の下流でmiR-199aが機能していることが明らかになった.
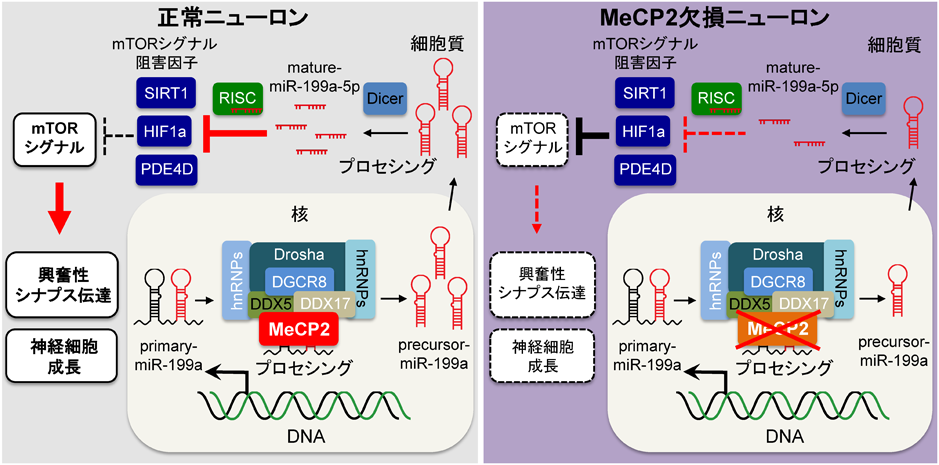
6)miR-199aの下流標的因子
以上のように,MeCP2はDrosha複合体の構成因子として特定miRNAのプロセシングを促進すること,MeCP2の下流標的miR-199aはMeCP2の下流で機能することが示された57)(図6).それではmiR-199aはMeCP2の下流でどのように神経機能を制御しているのであろうか? 筆者らはこの疑問に取り組むために,miR-199a下流標的因子の同定を試みた.いくつかのmiRNA標的検索データベースや文献検索により,mTORシグナルを制御することが報告されているmiR-199a標的因子候補を探索した.この探索により,phosphodiesterase 4d (Pde4d), sirtuin 1 (Sirt1), hypoxia-inducible factor α (Hif1α)のmRNAが3′-UTRに高度に保存されたmiR-199a標的配列を有することがわかった.これらの因子は先行研究により,mTORシグナルを負に制御することが報告されている60–62).miR-199aがこれらの因子を標的とするのかを調べるために,Pde4d, Sirt1, Hif1α 3′-UTRの野生型もしくは変異挿入型配列を含むレポーターコンストラクトを用いてルシフェラーゼアッセイを行った.
その結果,miR-199aの発現によりPde4d, Sirt1, Hif1αすべてのnativeレポーターコンストラクトのルシフェラーゼ活性の減少が認められた.一方,miR-199a標的配列に変異を持つmutantレポーターコンストラクトのルシフェラーゼ活性はmiR-199a発現による減弱を示さなかった.また,MeCP2KOマウス脳におけるPde4d, Sirt1, Hif1αのタンパク質量をイムノブロッティングにより調べたところ,有意な発現量の増加がみられた.さらに,MeCP2とPde4d, Sirt1, Hif1αそれぞれを野生型ニューロンに共発現させると,MeCP2発現により引き起こされるニューロン細胞体サイズ,興奮性シナプス伝達・形成の増加が消失した.これらの結果は,miR-199aがPde4d, Sirt1, Hif1αを標的としており,miR-199aを介したPde4d, Sirt1, Hif1αの発現制御がMeCP2の神経機能制御に重要であることを示している.
7)MeCP2下流におけるmTORシグナルの機能
これまでの先行研究により,MeCP2KOマウスや細胞において,mTORシグナル経路の制御異常が報告されていたが,mTORシグナルが実際にMeCP2の下流で機能しているのかは確かではなかった.そのため,筆者らはまず,mTORシグナルの活性化がMeCP2KOニューロンの表現型を改善できるのかを調べた.MeCP2KOニューロンにmTORシグナルの上流活性化因子であるRas homolog enriched in brain(Rheb)を発現させたところ,MeCP2KOニューロンにおいて観察される神経機能障害が野生型と同程度にまで回復した.次に,MeCP2を過剰発現するニューロンをmTORの阻害剤であるラパマイシンで処理したところ,MeCP2発現によって誘導される効果の消失がみられた.これらの結果は,ニューロンにおいて,mTORシグナルがMeCP2の下流で機能していることを示している(図7).
5. in vivoにおけるmiR-199aの機能
ここまでの結果より,MeCP2がmiR-199aを介してmTORシグナルを制御していることが明らかになったが,はたしてマウス個体においてもmiR-199aはRTT病態に関与しているのだろうか? 最後に筆者らは,in vivoにおけるmiR-199aの機能とRTT病態との関係を調べるために,miR-199aを欠損したマウスの作製を試みた.前述のように,miR-199aはゲノム上の二つの遺伝子座,miR-199a-1とmiR-199a-2にコードされる.miR-199a-1を欠損した(miR-199a-1KO)マウスは他グループによってすでに作製されており,miR-199a-1KOマウスは正常な発達を示し,明確な異常はみられないことが報告されている(http://jaxmice.jax.org/strain/017512.html).そのため,筆者らはmiR-199a-2を欠損する(miR-199a-2KO)マウスの作製を試みた.コンベンショナルな方法により,miR-199a-2遺伝子座のprecursor-miR-199aのみを欠失したマウスを作製した.驚いたことに,miR-199a-2KOマウスは,MeCP2KOマウスにおいて観察される多くのRTT表現型を示した.まず,miR-199a-2KOホモマウスは,生後3~4週齢ごろまでは一見正常な発達がみられるが,その後,野生型マウスと比較して全体の体の大きさ・体重および脳体積・重量に顕著な減少が観察される.一方,miR-199a-2KOヘテロマウスは野生型とほぼ同程度の発達を示した.また,miR-199a-2KOホモマウスは3~4週齢以降,低活動,歩行失調,イレギュラーな呼吸,振戦などのMeCP2KOマウスにみられる異常な表現型を示した.さらに免疫組織学的解析を行ったところ,miR-199a-2KOマウスは脳構造には異常はみられないが,MeCP2を正常に発現しているにも関わらず,海馬や大脳皮質ニューロンの細胞体が小さく,密にパックした配置を示すことが明らかになった.重要なことに,筆者らはmiR-199a-2KOマウスの海馬や大脳皮質などの脳領域において,MeCP2KOマウスと同様にmTORシグナルの顕著な減弱がみられることを突き止めた.以上の結果は,miR-199aがin vivoにおいてもmTOR活性を促進すること,MeCP2/miR-199a/mTORシグナルカスケードがRTT病態においてきわめて重要な役割を果たしていることを示している57).
RTTとその原因遺伝子産物であるMeCP2は,エピジェネティクスと脳機能・精神疾患を結びつける因子として脚光を浴び,世界中で精力的に研究がなされている.これまでの研究により,MeCP2は非常に多様な機能を駆使することにより中枢神経系の複雑かつ精巧な機能をつかさどっているように思われる.しかしながら,MeCP2の機能異常とRTT病態との関係はいまだ不明な点が多く残されている.近年,RTT表現型におけるグリア細胞でのMeCP2機能の重要性も示され,RTTの複雑な病態が浮き彫りになりつつある63, 64).一方で,RTT患者由来のiPS細胞を使った疾患研究65)や,MeCP2遺伝子重複霊長類モデルの登場26)により,MeCP2機能異常に起因する精神疾患研究の加速化が予想される.今後,さまざまなツールを用いたさらなるMeCP2分子機能の研究や下流標的因子の同定により,RTTを含めた精神・発達障害の病態解明および治療法開発が期待される.
謝辞Acknowledgments
本稿の執筆の機会を与えていただきました諸先生に深く感謝致します.また,本稿で紹介した筆者らの研究は,主に九州大学大学院医学研究院・応用幹細胞医科学部門・基盤幹細胞学分野にて行われたものですが,同時に多くの研究者との共同研究の成果でもあります.この場をかりて共同研究者の方々に感謝申し上げます.
引用文献References
1) de la Torre-Ubieta, L., Won, H., Stein, J.L., & Geschwind, D.H. (2016) Nat. Med., 22, 345–361.
2) Autism and Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators (2014) MMWR Surveill. Summ., 63, 1–21.
3) Rett, A. (1966) Wien. Med. Wochenschr., 116, 723–726.
4) Hagberg, B., Aicardi, J., Dias, K., & Ramos, O. (1983) Ann. Neurol., 14, 471–479.
5) Lewis, J.D., Meehan, R.R., Henzel, W.J., Maurer-Fogy, I., Jeppesen, P., Klein, F., & Bird, A. (1992) Cell, 69, 905–914.
6) Nan, X., Campoy, F.J., & Bird, A. (1997) Cell, 88, 471–481.
7) Amir, R.E., Van den Veyver, I.B., Wan, M., Tran, C.Q., Francke, U., & Zoghbi, H.Y. (1999) Nat. Genet., 23, 185–188.
8) Chahrour, M. & Zoghbi, H.Y. (2007) Neuron, 56, 422–437.
9) Takizawa, T., Nakashima, K., Namihira, M., Ochiai, W., Uemura, A., Yanagisawa, M., Fujita, N., Nakao, M., & Taga, T. (2001) Dev. Cell, 1, 749–758.
10) Namihira, M., Kohyama, J., Semi, K., Sanosaka, T., Deneen, B., Taga, T., & Nakashima, K. (2009) Dev. Cell, 16, 245–255.
11) Gottlicher, M. (2004) Ann. Hematol., 83(Suppl 1), S91–S92.
12) Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E., & Gage, F.H. (2004) Proc. Natl. Acad. Sci. USA, 101, 16659–16664.
13) Juliandi, B., Tanemura, K., Igarashi, K., Tominaga, T., Furukawa, Y., Otsuka, M.I., Moriyama, N., Ikegami, D., Abematsu, M., Sanosaka, T., Tsujimura, K., Narita, M., Kanno, J., & Nakashima, K. (2015) Stem Cell Rep., 5, 996–1009.
14) Christodoulou, J., Grimm, A., Maher, T., & Bennetts, B. (2003) Hum. Mutat., 21, 466–472.
15) Chen, R.Z., Akbarian, S., Tudor, M., & Jaenisch, R. (2001) Nat. Genet., 27, 327–331.
16) Guy, J., Hendrich, B., Holmes, M., Martin, J.E., & Bird, A. (2001) Nat. Genet., 27, 322–326.
17) Gemelli, T., Berton, O., Nelson, E.D., Perrotti, L.I., Jaenisch, R., & Monteggia, L.M. (2006) Biol. Psychiatry, 59, 468–476.
18) Guy, J., Gan, J., Selfridge, J., Cobb, S., & Bird, A. (2007) Science, 315, 1143–1147.
19) Giacomentti, E., Luikenhuis, S., Beard, C., & Jaenisch, R. (2007) Proc. Natl. Acad. Sci. USA, 104, 1931–1936.
20) McGill, B.E., Bundle, S.F., Yaylaoglu, M.B., Carson, J.P., Thaller, C., & Zoghbi, H.Y. (2006) Proc. Natl. Acad. Sci. USA, 103, 18267–18272.
21) Moretti, P., Bouwknecht, J.A., Teague, R., Paylor, R., & Zoghbi, H.Y. (2005) Hum. Mol. Genet., 14, 205–220.
22) Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R., & Zoghbi, H. (2002) Neuron, 35, 243–254.
23) Goffin, D., Allen, M., Zhang, L., Amorim, M., Wang, I.J., Reyes, A.S., Mercado-Berton, A., Ong, C., Cohen, S., Hu, L., Blendy, J.A., Carlson, G.C., Siegel, S.J., Greenberg, M.E., & Zhou, Z. (2012) Nat. Neurosci., 15, 274–285.
24) Lawson-Yuen, A., Liu, D., Han, L., Jiang, Z.I., Tsai, G.E., Basu, A.C., Picker, J., Feng, J., & Coyle, J.T. (2007) Brain Res., 1180, 1–6.
25) Collins, A.L., Levenson, J.M., Vilaythong, A.P., Richman, R., Armstrong, D.L., Noebels, J.L., Sweatt, J.D., & Zoghbi, H.Y. (2004) Hum. Mol. Genet., 13, 2679–2689.
26) Liu, Z., Li, X., Zhang, J., Cai, Y., Cheng, T., Chen, C., Wang, Y., Zhang, C., Nie, Y., Chen, Z., Bian, W., Zhang, L., Xiao, J., Lu, B., Zhang, Y., Zhang, X., Sang, X., Wu, J., Xu, X., Xiong, X., Zhang, F., Yu, X., Gong, N., Zhou, W., Sun, Q., & Qiu, Z. (2016) Nature, 530, 98–102.
27) Armstrong, D.D. (2005) J. Child Neurol., 20, 747–753.
28) Jellinger, K., Armstrong, D., Zoghbi, H.Y., & Percy, A.K. (1988) Acta Neuropathol., 76, 142–158.
29) Reiss, A.L., Faruque, F., Naidu, S., Abrams, M., Beaty, T., Bryan, R.N., & Moser, H. (1993) Ann. Neurol., 34, 227–234.
30) Belichenko, P.V., Oldfors, A., Hagberg, B., & Dahistrom, A. (1994) Neuroreport, 5, 1509–1513.
31) Moser, S.J., Weber, P., & Lutschg, J. (2007) Pediatr. Neurol., 36, 95–100.
32) Asaka, Y., Jugloff, D.G., Zhang, L., Eubanks, J.H., & Fitzsimonds, R.M. (2006) Neurobiol. Dis., 21, 217–227.
33) Moretti, P., Levenson, J.M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., Armstrong, D., Arancio, O., Sweatt, J.D., & Zoghbi, H.Y. (2006) J. Neurosci., 26, 319–327.
34) Dani, V.S., Chang, Q., Maffei, A., Turrigiano, G.G., Jaenisch, R., & Nelson, S.B. (2005) Proc. Natl. Acad. Sci. USA, 102, 12560–12565.
35) Nelson, E.D., Kavalali, E.T., & Monteggia, L.M. (2006) Curr. Biol., 16, 710–716.
36) Chao, H.T., Zoghbi, H.Y., & Rosenmund, C. (2007) Neuron, 56, 1–8.
37) Dragich, J.M., Kim, Y.H., Arnold, A.P., & Schanen, N.C. (2007) J. Comp. Neurol., 501, 526–542.
38) Kriaucionis, S. & Bird, A. (2004) Nucleic Acids Res., 32, 1818–1823.
39) Pelka, G.J., Watson, C.M., Christodoulou, J., & Tam, P.P. (2005) Genomics, 85, 441–452.
40) Kishi, N. & Macklis, J.D. (2004) Mol. Cell. Neurosci., 27, 306–321.
41) Klose, R.J., Sarraf, S.A., Schmiedeberg, L., McDermott, S.M., Stancheva, I., & Bird, A.P. (2005) Mol. Cell, 19, 667–678.
42) Buschdorf, J.P. & Stratling, W.H. (2004) J. Mol. Med. (Berl.), 82, 135–143.
43) Nan, X., Ng, H.H., Johnson, C.A., Laherty, C.D., Turner, B.M., Elsenman, R.N., & Bird, A. (1998) Nature, 393, 386–389.
44) Harikrishnan, K.N., Chow, M.Z., Baker, E.K., Pal, S., Bassal, S., Brasacchio, D., Wang, L., Craig, J.M., Jones, P.L., Sif, S., & El-Osta, A. (2005) Nat. Genet., 37, 254–264.
45) Kimura, H. & Shiota, K. (2003) J. Biol. Chem., 278, 4806–4812.
46) Horike, S., Cai, S., Miyano, M., Cheng, J.F., & Kohwi-Shigematsu, T. (2005) Nat. Genet., 37, 31–40.
47) Young, J.I., Hong, E.P., Castle, J.C., Crespo-Barreto, J., Bowman, A.B., Rose, M.F., Kang, D., Richman, R., Johnson, J.M., Berget, S., & Zoghbi, H.Y. (2005) Proc. Natl. Acad. Sci. USA, 102, 17551–17558.
48) Chahrour, M., Jung, S.Y., Shaw, S., Zhou, X., Wong, S.T., Qin, J., & Zoghbi, H.Y. (2008) Science, 320, 1224–1229.
49) Mellen, M., Ayata, P., Dewell, S., Kriaucionis, S., & Heintz, N. (2012) Cell, 151, 1417–1430.
50) Chen, W.G., Chang, Q., Lin, Y., Meissner, A., West, A.E., Griffith, E.C., Jaenisch, R., & Greenberg, M.E. (2003) Science, 302, 885–889.
51) Deng, V., Matagne, V., Banine, F., Freking, M., Ohliger, P., Budden, S., Pevsner, J., Dissen, G.A., Sherman, L.S., & Ojeda, S.R. (2007) Hum. Mol. Genet., 15, 640–650.
52) Itoh, M., Ide, S., Takashima, S., Kudo, S., Nomura, Y., Segawa, M., Kubota, T., Mori, H., Tanaka, S., Horie, H., Tanabe, Y., & Goto, Y. (2007) J. Neuropathol. Exp. Neurol., 66, 117–123.
53) Chang, Q., Khare, G., Dani, V., Nelson, S., & Jaenish, R. (2006) Neuron, 2, 341–348.
54) Costa-Mattioli, M. & Monteggia, L.M. (2013) Nat. Neurosci., 16, 1537–1543.
55) Ricciardi, S., Boggio, E.M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., Calcagno, E., Morello, N., Landsberger, N., Biffo, S., Pizzorusso, T., Giustetto, M., & Broccoli, V. (2011) Hum. Mol. Genet., 15, 1182–1196.
56) Li, Y., Wang, H., Muffat, J., Cheng, A.W., Orlando, D.A., Loven, J., Kwok, S.M., Feldman, D.A., Bateup, H.S., Gao, Q., Hockemeyer, D., Mitalipova, M., Lewis, C.A., Vander Heiden, M.G., Sur, M., Young, R.A., & Jaenisch, R. (2013) Cell Stem Cell, 3, 446–458.
57) Tsujimura, K., Ire, K., Nakashima, H., Egashira, Y., Fukao, Y., Fujiwara, M., Itoh, M., Uesaka, M., Imamura, T., Nakahata, Y., Yamashita, Y., Abe, T., Takamori, M., & Nakashima, K. (2015) Cell Reports, 22, 1887–1901.
58) Krol, J., Loedige, I., & Filipowicz, W. (2010) Nat. Rev. Genet., 11, 597–610.
59) Cheng, T.L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W.H., Xu, W., & Qiu, Z. (2014) Dev. Cell, 10, 547–560.
60) Kim, H.W., Ha, S.H., Lee, M.N., Huston, E., Kim, D.H., Jang, S.K., Suh, P.G., Houslay, M.D., & Ryu, S.H. (2010) Mol. Cell. Biol., 30, 5406–5420.
61) Ghosh, H.S., McBurney, M., & Robbins, P.D. (2010) PLoS ONE, 15, e9199.
62) Bruick, R.K. (2000) Proc. Natl. Acad. Sci. USA, 1, 9082–9087.
63) Lioy, D.T., Garg, S.K., Monaghan, C.E., Raber, J., Foust, K.D., Kaspar, B.K., Hirrlinger, P.G., Kirchhoff, F., Bissonnette, J.M., Ballas, N., & Mandel, G. (2011) Nature, 29, 497–500.
64) Derecki, N.C., Cronk, J.C., Lu, Z., Xu, E., Abbott, S.B., Guyenet, P.G., & Kipnis, J. (2012) Nature, 18, 105–109.
65) Marchetto, M.C., Carromeu, C., Acab, A., Yu, D., Yeo, G.W., Mu, Y., Chen, G., Gage, F.H., & Muotri, A.R. (2010) Cell, 12, 527–539.
著者紹介Author Profile
 辻村 啓太(つじむら けいた)
辻村 啓太(つじむら けいた)名古屋大学大学院医学系研究科神経情報薬理学講座特任助教.学位 博士(バイオサイエンス).
略歴1982年埼玉県に生る.2005年東京理科大学理学部卒業,07年奈良先端科学技術大学院大学バイオサイエンス研究科修士課程修了,10年同大学バイオサイエンス研究科博士課程修了,博士(バイオサイエンス)取得.10~13年同大学バイオサイエンス研究科博士研究員,13~15年九州大学大学院医学研究院応用幹細胞医科学部門基盤幹細胞学分野特任助教,15年より現職.
研究テーマと抱負発達障害・精神疾患の分子病態メカニズムの解明.疾患発症に重要な分子メカニズムを明らかにし,創薬へと発展するような研究を行っていきたい.
趣味自転車,温泉めぐり.
 中島 欽一(なかしま きんいち)
中島 欽一(なかしま きんいち)九州大学大学院医学研究院応用幹細胞医科学部門基盤幹細胞学分野教授.博士(理学).
略歴1967年熊本県に生る.90年九州大学理学部卒業,92年同大学院理学研究科修士課程修了,95年同大学院理学研究科博士課程修了,博士(理学)取得.日本学術振興会特別研究員として,大阪大学細胞生体工学センター,東京医科歯科大学難治疾患研究所を経たのち,98年東京医科歯科大学難治疾患研究所助手.2000年熊本大学発生医学研究センター助教授.02~04年米国ソーク研究所(日本学術振興会海外特別研究員).04年奈良先端科学技術大学院大学バイオサイエンス研究科教授.13年より現職.
研究テーマと抱負エピジェネティクスによる神経系細胞機能制御機構の解明.今後は基礎研究を行いながら,その成果・知見に立脚した,再生医療への応用も見据えた研究を行いたい.
ウェブサイトhttps://scb.wp.med.kyushu-u.ac.jp/
趣味ギター (Hard Rock).