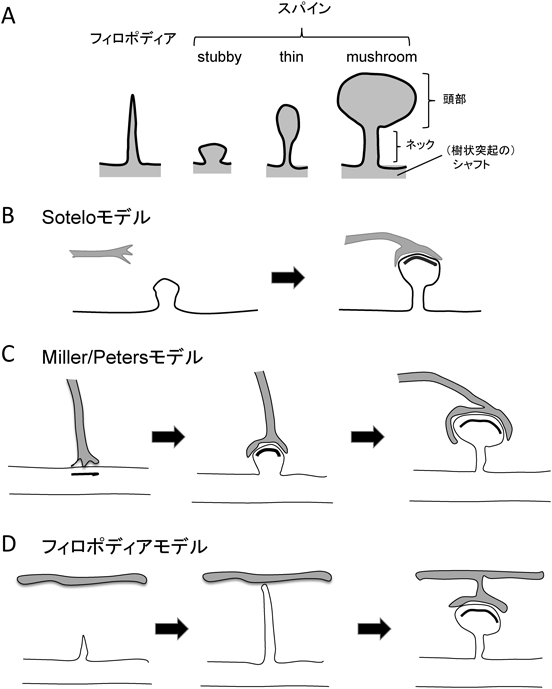
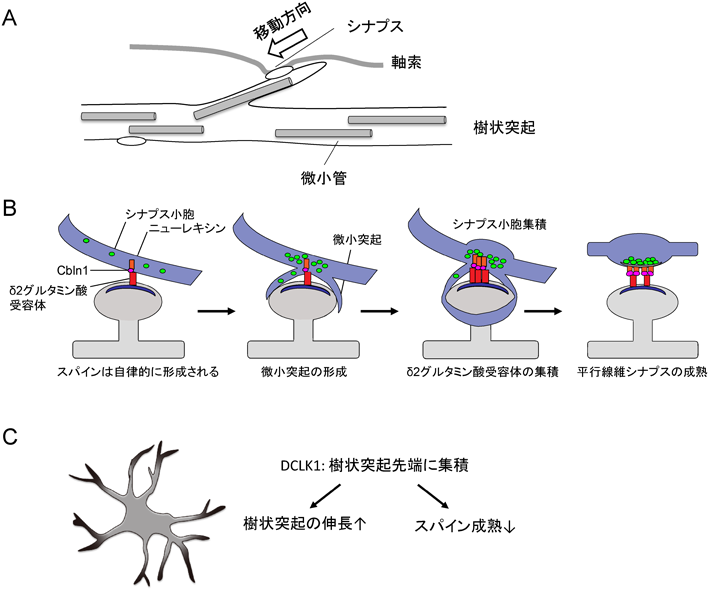
中枢神経系シナプス形成の多様性の分子的基盤Diversity of the molecular mechanisms of synapse formation in the central nervous system
東京大学大学院医学系研究科神経細胞生物学分野Department of Cellular Neurobiology, Graduate School of Medicine, The University of Tokyo ◇ 〒113–0033 東京都文京区本郷7–3–1 ◇ 7–3–1 Hongo, Bunkyo-ku, Tokyo 113–0033, Japan
発行日:2017年2月25日Published: February 25, 2017