3) Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, J.P., Deng, H.X., Rahmani, Z., Krizus, A., McKenna-Yasek, D., Cayabyab, A., Gaston, S.M., Berger, R., Tanzi, R.E., Halperin, J.J., Herzfeldt, B., den Bergh, R.V., Hung, W.-Y., Bird, T., Deng, G., Mulder, D.W., Smyth, C., Laing, N.G., Soriano, E., Pericak-Vance, M.A., Haines, J., Rouleau, G.A., Gusella, J.S., Horvitz, H.R., & Brown, R.H. Jr. (1993) Nature, 362, 59–62.
4) Iuchi, Y., Okada, F., Onuma, K., Onoda, T., Asao, H., Kobayashi, M., & Fujii, J. (2007) Biochem. J., 402, 219–227.
5) Iuchi, Y., Okada, F., Takamiya, R., Kibe, N., Tsunoda, S., Nakajima, O., Toyoda, K., Nagae, R., Suematsu, M., Soga, T., Uchida, K., & Fujii, J. (2009) Biochem. J., 422, 313–320.
7) Iuchi, Y., Kibe, N., Tsunoda, S., Suzuki, S., Mikami, T., Okada, F., Uchida, K., & Fujii, J. (2010) Free Radic. Biol. Med., 48, 935–944.
8) Konno, T., Otsuki, N., Kurahashi, T., Kibe, N., Tsunoda, S., Iuchi, Y., & Fujii, J. (2013) Free Radic. Biol. Med., 65, 1378–1384.
10) Otaki, N., Chikazawa, M., Nagae, R., Shimozu, Y., Shibata, T., Ito, S., Takasaki, Y., Fujii, J., & Uchida, K. (2010) J. Biol. Chem., 285, 33834–33842.
11) Otsuki, N., Konno, T., Kurahashi, T., Suzuki, S., Lee, J., Okada, F., Iuchi, Y., Homma, T., & Fujii, J. (2016) Free Radic. Res., 50, 793–800.
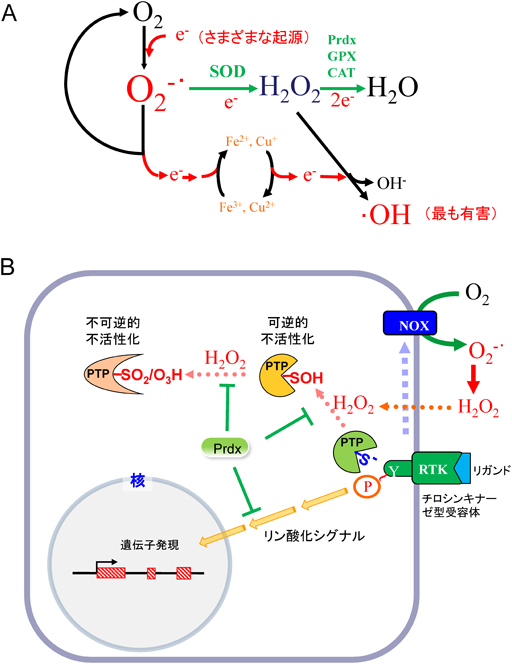
12) Lee, T.H., Kim, S.U., Yu, S.L., Kim, S.H., Park, D.S., Moon, H.B., Dho, S.H., Kwon, K.S., Kwon, H.J., Han, Y.H., Jeong, S., Kang, S.W., Shin, H.S., Lee, K.K., Rhee, S.G., & Yu, D.Y. (2003) Blood, 101, 5033–5038.
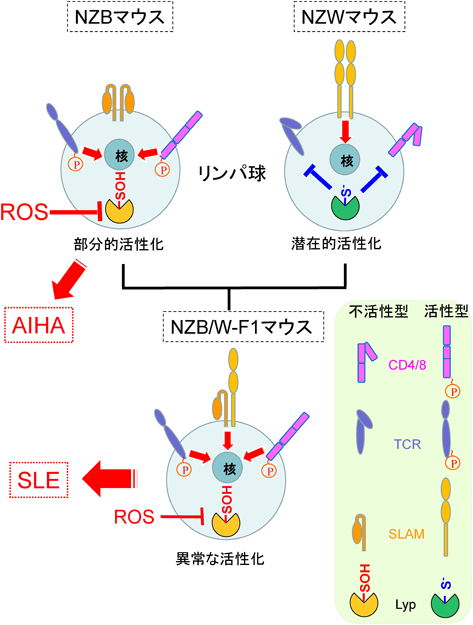
14) Wandstrat, A.E., Nguyen, C., Limaye, N., Chan, A.Y., Subramanian, S., Tian, X.H., Yim, Y.S., Pertsemlidis, A., Garner, H.R. Jr., Morel, L., & Wakeland, E.K. (2004) Immunity, 21, 769–780.