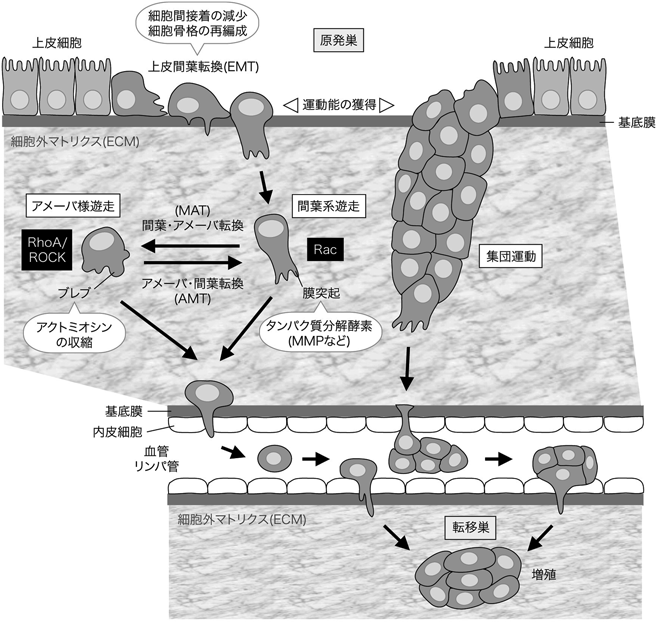
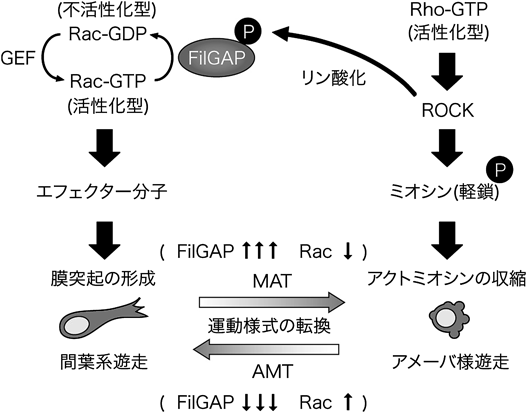
がん細胞の浸潤における運動様式の転換制御Control of switching between migration modes in cancer invasion
北里大学理学部生物科学科細胞生物学講座Division of Cell Biology, Department of Biosciences, School of Science, Kitasato University ◇ 〒252–0373 神奈川県相模原市南区北里1–15–1 ◇ 1–15–1 Kitasato, Sagamihara, Minami-ku, Kanagawa 252–0373, Japan
発行日:2017年2月25日Published: February 25, 2017