ゴルジ体ストレス応答Golgi stress response
兵庫県立大学大学院生命理学研究科Graduate School of Life Science, University of Hyogo ◇ 〒678–1297 兵庫県赤穂郡上郡町光都3–2–1 ◇ 3–2–1 Koto, Harima Science Garden City, Hyogo 678–1297, Japan
発行日:2017年4月25日Published: April 25, 2017
細胞小器官の存在量は,細胞の需要に応じて厳密に制御されている.特定の細胞小器官の機能が不足すると,特定の細胞小器官だけが必要な量だけ増強される.このような細胞小器官の量的調節機構は細胞が自律的に機能するために必須な機構であり,細胞生物学の重要問題である.ゴルジ体ストレス応答はゴルジ体の量的調節機構であり,この重要な問題を解明するための端緒となる研究である.哺乳類のゴルジ体ストレス応答経路にはTFE3経路とCREB3経路,HSP47経路がある.TFE3経路はゴルジ体の糖鎖修飾酵素や構造タンパク質,小胞輸送因子の発現を調節している.一方,CREB3経路はゴルジ体ストレスによるアポトーシスを誘導し,HSP47経路はアポトーシスを抑制している.ゴルジ体は複雑であるため,この3経路以外にも応答経路が存在する可能性が大きい.
© 2017 公益社団法人日本生化学会© 2017 The Japanese Biochemical Society
2016年10月,大隅良典博士がオートファジーの研究でノーベル医学・生理学賞を受賞した.大隅博士の「人がやらないことをやる」という言葉が心に沁みた.志の高さは天と地ほどの差があるが,筆者もまったく同感である.新しい生命現象を自分で見つけ,その機構や生理的重要性を研究することこそ,研究の醍醐味であり,研究者の存在意義である.そう考えて,無謀にも「細胞小器官の量的調節機構」の研究を志し,その端緒にゴルジ体の量的調節機構であるゴルジ体ストレス応答(Golgi stress response)の解析を行っている.最初に,細胞小器官の量的調節機構の重要性について述べる.
細胞の中には核やミトコンドリア,小胞体,ゴルジ体,リソソーム,ペルオキシソームのような細胞小器官が存在し,細胞の機能を分担している.同じタイプの細胞であれば,ほぼ同じ量の細胞小器官のセットが存在する.細胞が分裂する際,これらの細胞小器官の量はほぼ半分になる.細胞内の核の量は厳密に制御されており,通常の細胞では常に1個の核が存在するようになっているが,その他の細胞小器官の量はどうであろうか? 細胞分裂によって他の細胞小器官の量もほぼ半分になるが,細胞の成長とともにほぼ2倍に増やされるようである(図1A).細胞分裂に伴って小胞体がなくなったり,逆に細胞がゴルジ体だらけになったりしてしまうようなことはない.したがって,核以外の細胞小器官の量も厳密に制御されているはずである.
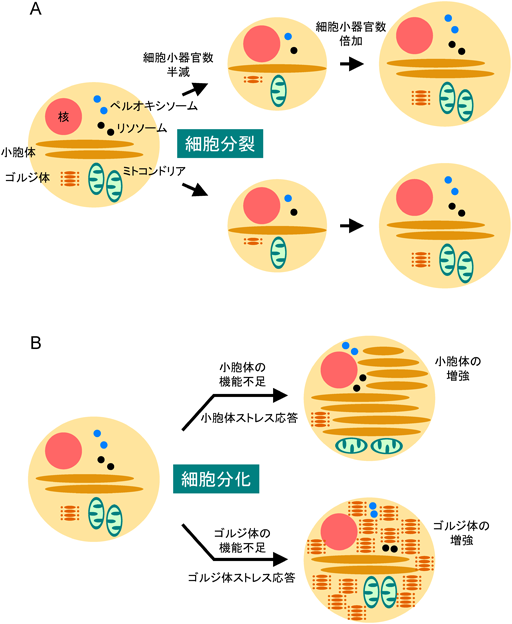
(A)細胞が分裂・成長する際の量的調節:細胞が分裂すると細胞小器官の量はほぼ半分になるが,細胞の成長に伴って倍加するため,量は一定に保たれる.(B)細胞が分化する際の量的調節:細胞が分化して特定の細胞小器官の量が不足すると,特定の細胞小器官の量が特異的に増強される.
細胞小器官の量を一定に保つ機構があると述べたが,細胞が置かれている状況が変化すれば,それに応じて細胞小器官の量はダイナミックに増減する.たとえば,抗体産生細胞の前駆体細胞には小胞体は少量しか存在しないが,病原菌が感染して前駆体細胞が抗体産生細胞に分化すると小胞体が増産され,細胞質が小胞体で埋めつくされる.抗体産生細胞は大量の抗体タンパク質を産生する必要があるので,抗体タンパク質を作る小胞体が増量されるのは理にかなっている.このように,特定の細胞小器官の機能が不足したときには,必要な細胞小器官だけが必要な量だけ増量される.このようなメカニズムがあれば,細胞分裂によって細胞小器官が減っても,ちゃんと元どおりに増やすことができる(図1B).このような細胞小器官の量的調節機構(organelle autoregulation)は細胞の需要に応じて細胞小器官の量を自動的に調節する機構であり,真核細胞が自律的に機能するために必須な機構である.細胞小器官の量的調節機構の問題は細胞生物学の根幹に関わる重大な研究課題であるにも関わらず,これまでは「ミトコンドリアや小胞体などというものは,適当に増えていくもの」として,核の量的調節機構(細胞周期の研究)以外はほとんど顧みられることがなかった.
ゴルジ体ストレス応答の先行研究として,小胞体ストレス応答がある.小胞体ストレス応答は小胞体の量的調節機構であると筆者は考えるが,この考え方は一般的ではなく,小胞体ストレス応答は小胞体ストレスという危機的状態に対抗するための生体防御機構と考えるのが一般的であった.少なくとも,すべての細胞小器官にそれぞれ量的調節機構があるという概念は存在しなかった.そこで,最初にゴルジ体にも量的調節機構(ゴルジ体ストレス応答)が存在することを証明し,その分子機構を明らかにすることによって,「細胞小器官の量的調節機構」という新しい研究分野を開拓することにした.
それでは,ゴルジ体の量的調節機構(ゴルジ体ストレス応答)とは,どのようなものか? ゴルジ体にはいろいろな機能があるが,主たる機能は翻訳後修飾(糖鎖修飾など)と小胞輸送である.ゴルジ体にさまざまな翻訳後修飾酵素や小胞輸送因子が存在し,分泌タンパク質や膜タンパク質の翻訳後修飾と小胞輸送を行っている(図2A).細胞分化によってタンパク質合成が増加するとどうなるか? 小胞体で大量の分泌タンパク質が合成されてゴルジ体にやってきたら,翻訳後修飾酵素が不足して翻訳後修飾が行えなくなるし,小胞輸送因子が不足するために細胞膜まで運ぶこともできなくなる(図2B).そこで,細胞はこのようなゴルジ体の機能不足(ゴルジ体ストレス状態)に対処するべく,ゴルジ体ストレス応答を活性化することによって翻訳後修飾酵素や小胞輸送因子の遺伝子の転写を増加させ,ゴルジ体の機能を増強する(図2C).これこそが,ゴルジ体ストレス応答(ゴルジ体の量的調節機構)である.
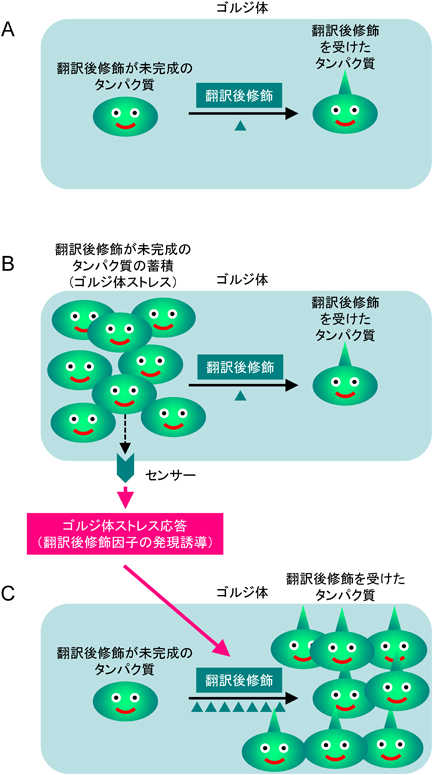
翻訳後修飾因子だけに注目して描画してあるが,小胞輸送因子についても同様である.
では,いったい個体のどこでゴルジ体ストレス応答は起こっているのか? 最も顕著な例は,消化管などにみられる粘膜分泌細胞の分化過程である.粘膜の主成分はムチンと呼ばれる糖タンパク質であり,コアとなるタンパク質に対してたくさんのO型糖鎖がブラシの毛のように付加されている.このO型糖鎖の付加反応はゴルジ体で起こるため,ムチンを産生する細胞(ゴブレット細胞など)ではゴルジ体が非常に発達している.このようなムチン産生細胞ではおそらくゴルジ体ストレス応答が顕著に活性化しているものと考えられる.もちろん,通常の細胞でもゴルジ体ストレス応答は常に機能しており,細胞の需要に応じてゴルジ体の量を調節していると考えている.
後述するように,哺乳類のゴルジ体ストレス応答は,TFE3経路1)とHSP47経路2),CREB3経路3)という三つの応答経路が知られている(図3).最初に,我々が同定したTFE3経路について述べる.
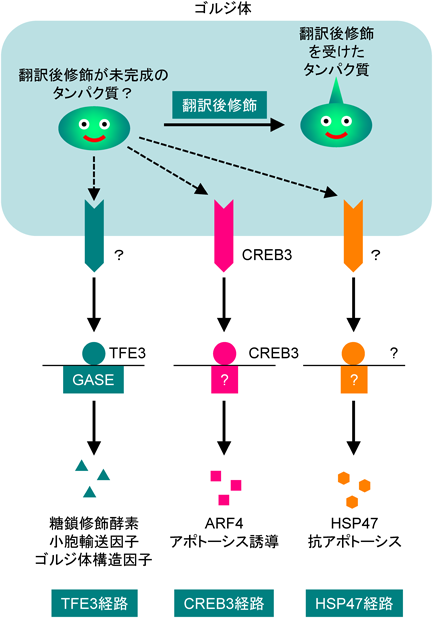
糖鎖修飾酵素や小胞輸送因子の発現を制御するTFE3経路と,アポトーシスを制御するHSP47経路とCREB3経路が存在する.ただし,センサーや転写因子などわかっていないことも多い.
ゴルジ体ストレス応答を実験的に解析するためには,まず人工的にゴルジ体ストレス(ゴルジ体の機能が不足する状態)を誘導する方法を確立する必要がある.小胞体ストレスの場合は,thapsigarginやtunicamycin, dithiothreitol, A23187などの薬剤を用いて小胞体ストレス(小胞体の機能が不足する状態)を誘導している4).このうちthapsigarginは小胞体にCa2+イオンを取り込ませるCa2+ポンプの阻害剤であり,A23187はCa2+イオンを小胞体から流出させるイオノフォアであるが,どちらも小胞体に高濃度で存在するCa2+イオンを減少させることで小胞体シャペロンの機能を低下させ(小胞体シャペロンの活性には高濃度のCa2+イオンが必要),小胞体ストレスを誘導している.一方,ゴルジ体は酸性の細胞小器官であり,ゴルジ体のタンパク質(翻訳後修飾酵素や小胞輸送因子)は酸性下で機能するようになっている.そこで,monensinやnigericinのようなイオノフォアで処理することによってH+イオンを流出させてゴルジ体内を中性化し,ゴルジ体のタンパク質の機能を低下させることでゴルジ体ストレス(ゴルジ体機能が不足する状態)を起こすことにした5).monensinで細胞を処理すると糖鎖修飾や小胞輸送が阻害されること,またゴルジ体が断片化して細胞中にゴルジ体が充満することが知られている6).このように,monensinやnigericinはゴルジ体機能の強い阻害剤であり,ゴルジ体ストレス誘導剤として有用なものである.
ただし,リソソームも酸性であることから,monensinはゴルジ体だけでなくリソソームの機能も不足させる可能性がある.しかしながら,monensin処理した細胞のマイクロアレイ解析では,リソソーム関連の遺伝子はmonensinによってほとんど転写誘導されなかった(図4).これは,リソソームとゴルジ体の膜脂質の成分が異なることから,monensinがゴルジ体膜に比較的選択的に挿入されることが原因と考えられている6).

また,monensinやnigericin以外にゴルジ体ストレス応答を活性化させる方法も検討した.タンパク質のフォールディングを阻害することで小胞体ストレスを起こすように,ゴルジ体での糖鎖修飾を阻害することでゴルジ体ストレス応答の誘導を試みた.ゴルジ体では糖鎖修飾の一種であるシアル酸修飾が起こるが,シアル酸修飾の材料であるCMP-シアル酸はSLC35A1と呼ばれるトランスポーターによって細胞質からゴルジ体へと運ばれる7).したがって,SLC35A1の発現をRNA干渉法によって抑制すれば,ゴルジ体でのシアル酸修飾を阻害することが可能である8).このように,シアル酸修飾を阻害することによってもゴルジ体ストレスを誘導することができる1).
さらに,ゴルジ体の構造タンパク質であるGCP60のドミナントネガティブ変異体(GCP60-DN)5)を過剰発現することによっても,ゴルジ体ストレスを誘導することができる.ゴルジ体ストレス応答によって発現が誘導されるGCP60はゴルジ体の表在タンパク質であり,C末端側領域でGiantinと相互作用することでゴルジ体の構造を維持したり,ゴルジ体以降の小胞輸送を制御したりしていると考えられている.GCP60のN末端側領域を欠失したGCP60-DNは過剰発現するとドミナントネガティブ変異体として作用し,ゴルジ体の構造を破壊し,小胞輸送機能を阻害することが報告されている9).そこで,GCP60-DNを過剰発現してゴルジ体の機能が不足する状況を作ったところ,確かにゴルジ体ストレス応答が活性化されることがわかった1, 5).
次に行ったのは,TFE3経路によって発現が誘導される標的遺伝子の検索である.細胞をゴルジ体ストレス誘導剤であるmonensinで処理し,マイクロアレイ解析や次世代DNAシークエンサー解析,qRT-PCRアレイ解析を行ったところ,monensin処理によって転写が誘導されるゴルジ体関連遺伝子を同定することができた(図4)5).これらの標的遺伝子を大まかに分類すると,3グループに分けられる.第一のグループは,ゴルジ体に存在する翻訳後修飾酵素である.これらのうちSIAT4AとSIAT10Aはシアル酸の転移酵素であり,FUT1はフコースの転移酵素,B3GAT3はグルクロン酸の転移酵素,UAP1L1は糖鎖付加の原材料であるUDP-N-アセチルグルコサミンを作る酵素である.PCSK1は,インスリンなどの分泌タンパク質のプロセシングを行うプロテアーゼである.ゴルジ体は小胞体で合成された分泌タンパク質や膜タンパク質の翻訳後修飾を行うところであり,これらの翻訳後修飾酵素の発現がゴルジ体ストレス時(ゴルジ体の機能が不足するとき)に誘導されることは理にかなっている.
第二のグループは,ゴルジ体以降の小胞輸送に関わる因子である.STX3Aは主として細胞膜に局在するt-SNAREであり,ゴルジ体から細胞膜への小胞小胞輸送を制御している.WIPI49はゴルジ体以降の膜輸送に関与しており,オートファジーにも関与している.RAB20はゴルジ体以降の小胞輸送に制御する低分子Gタンパク質であるが,その機能の詳細は不明である.小胞輸送もゴルジ体の重要な機能であり,ゴルジ体ストレス時にこれらの小胞輸送因子群の発現を誘導して小胞輸送能力を高めることも理にかなっている.
第三のグループは,ゴルジ体の構造タンパク質である.GCP60はゴルジ体の表在タンパク質であり,Giantinなどと結合することでゴルジ体の構造を維持したり,小胞輸送の足場になったりすると考えられている.GiantinやGM130はコイルドコイル構造を持つGolginファミリーに属するタンパク質であり,GCP60と同じくゴルジ体以降の小胞輸送を制御していると考えられている.ゴルジ体ストレス時には,このようなゴルジ体の構造タンパク質の発現を誘導することによってゴルジ体の土台を増やしているものと考えられる.
これらのTFE3経路の標的遺伝子は,thapsigarginのような小胞体ストレス誘導剤によっては発現誘導を受けなかった.一方,BiPやEDEM, CHOPのような小胞体ストレス応答の標的遺伝子の発現はmonensinによって影響を受けず,小胞体ストレス応答経路であるATF6経路やXBP1経路もmonensinによって活性化されないことから,ゴルジ体ストレス応答のTFE3経路と小胞体ストレス応答はまったく別個の制御機構であることがわかった.
これらmonensinによって転写が誘導される標的遺伝子のうち,SIAT4遺伝子とGCP60遺伝子のプロモーターの欠失変異体や点変異体の解析から,monensin処理による転写誘導を制御するエンハンサー配列を同定し,そのコンセンサス配列がACG TGG Cであることを明らかにした.この配列は,標的遺伝子群のプロモーター領域に共通してみられることから,Golgi apparatus stress response element(GASE)と呼ぶことにした5)(図3).
酵母細胞を用いたワンハイブリッド法によってGASEに結合して転写を制御する因子を検索したところ,二つの転写因子TFE3とMLXを同定した1).どちらもbasic helix-loop-helix leucine zipper(bHLH-ZIP)型転写因子に属する転写因子であるが,TFE3には強力な転写活性化領域が存在し,MLXには見当たらない(図5).TFE3を過剰発現したところGASEからの転写が上昇し,TFE3の発現を抑制するとゴルジ体ストレス時にみられるGASEからの転写誘導が低下したことから,TFE3は活性化型の転写因子であると考えられる.TFE3に対する抗体を作製して細胞を染色したところ,平常時にはTFE3は細胞質に存在しているが,monensin処理によってゴルジ体ストレスを与えると,3時間ほどで核へ移行することがわかった.ウエスタンブロットを行って分子量を調べたところ,平常時にはTFE3は高度にリン酸化されており,ゴルジ体ストレス時には脱リン酸化されることがわかった.リン酸化部位になりうるセリン・トレオニン残基を一つずつアラニン残基に置換したところ,108番目のセリン残基(Ser108)を置換するとTFE3は常に脱リン酸化状態になり,恒常的に核に存在することがわかった.これらのことから,TFE3は平常時にはSer108がリン酸化されることによって細胞質に繋留されており,ゴルジ体ストレス時にSer108が脱リン酸化されることで核へ移行していると考えている.TFE3のリン酸化状態を制御するリン酸化酵素や脱リン酸化酵素は,現在検索中である.
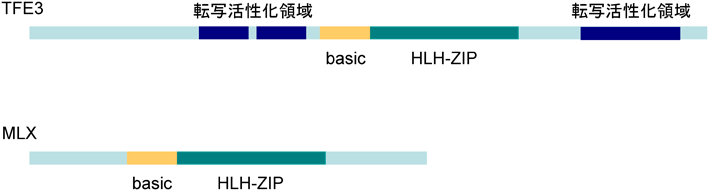
どちらもbasic-helix-loop-helix leucine zipper(bHLH-ZIP)を持つ転写因子だが,MLXには転写活性化領域が見当たらない.TFE3は転写活性化因子,MLXは転写抑制因子であると考えている.
一方,MLXを細胞で過剰発現すると,ゴルジ体ストレス時のGASEからの転写誘導が低下し,MLXの発現を抑制すると転写誘導は逆に上昇した10).このことは,MLXがTFE3経路を負に制御する転写制御因子であることを示している.TFE3を過剰発現したときにMLXを共発現させると,TFE3によるGASEからの転写誘導が低下することから,MLXはTFE3の作用を阻害していることがわかった.そこでTFE3とMLXのGASEへの結合をみると,MLXはTFE3と競合的にGASEに結合することで,GASEからの転写誘導を阻害していることがわかった.MLXの細胞内局在性を見たところ,TFE3と同様に平常時は細胞質に繋留されており,ゴルジ体ストレス時には核へ移行することがわかった.MLXが細胞質に繋留されているメカニズムはまだわかっていない.
現在わかっているTFE3経路の制御メカニズムを図6に示す.平常時にはTFE3もMLXも細胞質にとどめられており,標的遺伝子の転写誘導は起こらない.ゴルジ体ストレス時にはTFE3が核移行してGASEに結合し転写を誘導しようとするが,負の制御因子であるMLXも核移行してGASEを奪い合うことで転写誘導を止めようとする.このような二つの相反する応答の微妙なバランスによって転写誘導の度合いが決められているものと想像される.小胞体ストレス応答を制御する転写因子ATF6もこれとよく似ている.ATF6にはATF6αとATF6βの二つの遺伝子が存在し,ATF6αは強力な転写誘導能を持つのに対して,ATF6βは弱い転写誘導能しか持っていない.ATF6βを細胞内で過剰発現するとATF6βがエンハンサーであるERSEに結合してしまい,ATF6αがERSEに結合するのを邪魔してしまうため,ERSEからの転写誘導は低下してしまう.ATF6経路もこの二つの転写因子の絶妙なバランスによって転写誘導の度合いが制御されている.

TFE3もMLXも,平常時は細胞質に不活性な状態で繋留されているが,ゴルジ体ストレス時には両者が核へ移行してGASEを奪い合い,両者のバランスによって標的遺伝子の転写量が決定される.
遠山正彌博士(大阪大学)と宮田信吾博士(近畿大学)のグループは,ゴルジ体ストレス応答のもう一つの経路としてHSP47経路を報告している2)(図3).HSP47経路は,糖鎖修飾酵素ではなく小胞体シャペロンであるHSP47の発現を誘導することによって,ゴルジ体ストレスによる細胞死を抑制する経路である.彼らはムチン型糖鎖修飾の阻害剤であるbenzyl-2-acetamido-2-deoxy-α-D-galactopyranoside(BG)を用いてゴルジ体ストレス応答を起こしている.ムチン型糖鎖修飾はコアタンパク質のセリン・トレオニン残基にN-アセチルガラクトサミン(GalNAc)が結合することによって開始される.細胞中にGalNAcの誘導体であるBGが多量に存在すると,二つ目以降の糖がコアタンパク質に結合したGalNAcではなくBGに結合してしまうため,コアタンパク質のムチン型糖鎖修飾が競合的に阻害される.ムチン型糖鎖修飾能力を不足させたときにゴルジ体ストレス応答が起こるかどうか調べるために,ムチン型糖鎖修飾が盛んなヒト大腸がん細胞であるColo 205細胞をBG処理し,細胞から抽出したタンパク質を二次元電気泳動によって解析したところ,BG処理によってHSP47の発現が著しく上昇することがわかった.この発現誘導はHSP47遺伝子の転写レベルでの誘導である.
HSP47は永田和宏博士(京都大学・京都産業大学)が発見したコラーゲン専用の分子シャペロンであり,小胞体内でのコラーゲンのプロリン残基水酸化や三本鎖形成などの成熟過程に重要な役割を果たしている.HSP47の発現は熱ショックによって誘導されることが知られているが,tunicamycinやthapsigarginのような小胞体ストレス誘導剤によっては誘導されない.興味深いことに,ゴルジ体ストレス誘導剤であるmonensinによってもHSP47の発現が誘導される.HSP47は小胞体に局在していることが知られており,BG処理をしても小胞体への局在は変化しない(ゴルジ体には移行しない).
HSP47の発現を抑制すると,BG処理によってゴルジ体が拡張・分断されている像がみられ,またゴルジ体に局在するカスパーゼであるカスパーゼ-2が活性化されて細胞死を起こしやすくなる.HSP47を過剰発現しておくと細胞死が抑制されることから,ゴルジ体ストレス時にはHSP47遺伝子の転写が誘導され,ゴルジ体ストレスによる細胞死から細胞を守っていると考えられる.BG処理だけでは小胞体ストレス応答は活性化されないが,HSP47の発現を抑制してBG処理をするとpATF6(P)(pはタンパク質を表し,Pはprecursorの意味である)の切断やCHOPの発現誘導が起こることから,小胞体ストレスによる細胞死が誘導されている可能性もある.コラーゲンの発現には影響がなかった.
ムチン型糖鎖修飾が起こるのはゴルジ体であり,一方HSP47が存在・機能するのは小胞体である.なぜゴルジ体ストレスによってHSP47の発現が誘導され,細胞死を抑制できるのか,その機構は不明である.コラーゲンはゴルジ体でムチン型糖鎖修飾を受けることから,ゴルジ体へ一時的に運ばれたHSP47がゴルジ体でコラーゲンの成熟を促進している可能性もある.HSP47が不足すると,ゴルジ体でのコラーゲンの成熟が阻害され,それをゴルジ体ストレスのセンサーが感知することによってカスパーゼ-2が活性化されて細胞死が起こるのかもしれない.あるいは,ゴルジ体でムチン型糖鎖修飾を受けたタンパク質が小胞体へ移行し機能する例も知られていることから,そのようなタンパク質がムチン型糖鎖修飾を受けずに小胞体へ運ばれ,小胞体ストレスを起こすことで細胞死が誘導されている可能性も考えられる.また,HSP47の発現がゴルジ体ストレスによって誘導される分子機構も,今後の重要な研究課題である.
David Sabatini博士(MIT)は,ゴルジ体ストレス応答のCREB3経路について報告している3)(図3).CREB3経路は,小胞輸送を制御する低分子Gタンパク質ARF4の発現を誘導することによって,ゴルジ体ストレスによる細胞死を促進する経路である.彼らが用いているゴルジ体ストレス誘導剤は,brefeldin A(BFA)である.BFAはもともと抗ウイルス用の抗生物質であり,その作用点は低分子Gタンパク質であるARFである.BFAはARFに結合することで小胞体からゴルジ体への輸送を阻害するため,ゴルジ体はやがて小胞体へ吸収される.BFAによって細胞死が起こることは以前から知られていたが,その過程に関与する因子を同定するために,彼らはほぼ一倍体であるヒト慢性骨髄性白血病細胞由来KBM細胞に挿入変異を行い,BFAによる細胞死に耐性であるクローンをスクリーニングした.次世代シークエンサーを用いてゲノム配列を解析し挿入変異によって破壊された遺伝子を調べたところ,ARF4を同定した.ARF4の発現を抑制すると,BFAだけでなく他のゴルジ体ストレス誘導剤であるGolgicide A(ARFのグアニンヌクレオチド交換因子であるGBF1の阻害剤)やExo1(ARFの阻害剤)による細胞死が軽減されたが,小胞体ストレスによる細胞死には影響がみられなかった.また,ARF4の発現を抑制しておくと,BFAで処理してもタンパク質の糖鎖修飾や分泌は阻害されず,ゴルジ体が小胞体に吸収されることもなくなった.興味深いことに,BFA処理によってARF4の発現が誘導されたが,小胞体ストレスによっては影響を受けなかった.彼らは,このゴルジ体ストレスによるARF4遺伝子の転写誘導を制御している転写因子がCREB3であることを明らかにした.CREB3はATF6ファミリーに属する転写因子で,平常時は小胞体膜上に膜貫通型タンパク質として存在するが,BFAやGolgicide A, Exo1, monensin処理のようなゴルジ体ストレスによって小胞体からゴルジ体へ移行し,ゴルジ体に存在するプロテアーゼS1PとS2Pによって切断を受けて膜から遊離し,核へ移行して転写を誘導した.このようにARF4はゴルジ体ストレスによる細胞死を促進する機能を持つが,ARF1やARF5は逆にゴルジ体ストレスによる細胞死を抑制する機能を持っていた.
CREB3は別名Lumanとも呼ばれ,ATF6と同様に小胞体ストレスによって活性化されることが知られている.BFAはゴルジ体を小胞体に吸収させるためにゴルジ体に存在しているS1PやS2Pを小胞体へ移行させ,その結果ATF6やCREB3が切断されることも知られており,BFA処理時にCREB3が切断されることがゴルジ体ストレスによるものなのか,ゴルジ体が小胞体に吸収されたためなのか判別が難しい.また,小胞体に存在するCREB3がどのようにしてゴルジ体ストレスを感知できるのか,その分子機構は今後の重要な研究課題である.
ゴルジ体以外の細胞小器官の量的調節機構も最近続々と報告されるようになっている.以下に,その概要を記載する.
線虫(Caenorhabditis elegans)のミトコンドリアの量的調節機構はmitochondrial unfolded protein responseと呼ばれ,ATFS-1経路11)とROS-GCN2経路12)という2種類の応答経路が知られている(図7).ミトコンドリア内でも細胞質と同じようにタンパク質のフォールディングが行われており,ミトコンドリアでのタンパク質合成が増加するとミトコンドリアのシャペロンが不足する状況(ミトコンドリアストレス状態)となって,正常な立体構造を形成していないタンパク質が蓄積する.これらの構造異常タンパク質はAAA-ATPaseファミリーに属するプロテアーゼであるCLPP1によって分解されてペプチド断片となり13),このペプチド断片がミトコンドリア膜上に存在するペプチドトランスポーターHAF-1によって細胞質へと輸送され14),転写因子DVE1やATFS-1を活性化すると考えられている.転写因子ATFS-1は核移行シグナルとミトコンドリア移行シグナルの両方を持っているが,平常時はミトコンドリア移行シグナルの方が強いために,ミトコンドリア内(マトリクス内)に輸送され,プロテアーゼによって分解されている15).ミトコンドリアストレス時には,細胞質に輸送されたペプチド断片がATFS-1のミトコンドリアへの輸送を阻害し(詳細なメカニズムは不明),その結果ATFS-1は核へ移行してミトコンドリアのシャペロンや不良品を分解するプロテアーゼの発現を転写レベルで誘導する.また,ATFS-1は核ゲノムやミトコンドリアゲノムに存在する酸化的リン酸化に関与する遺伝子の転写を抑制することで,ミトコンドリアストレスを軽減しようとする16).ATFS-1経路はミトコンドリアストレス応答を介して,神経変性疾患の抑制17)や細胞の老化18),寿命19, 20),自然免疫21)などさまざまな生命現象に関与している.
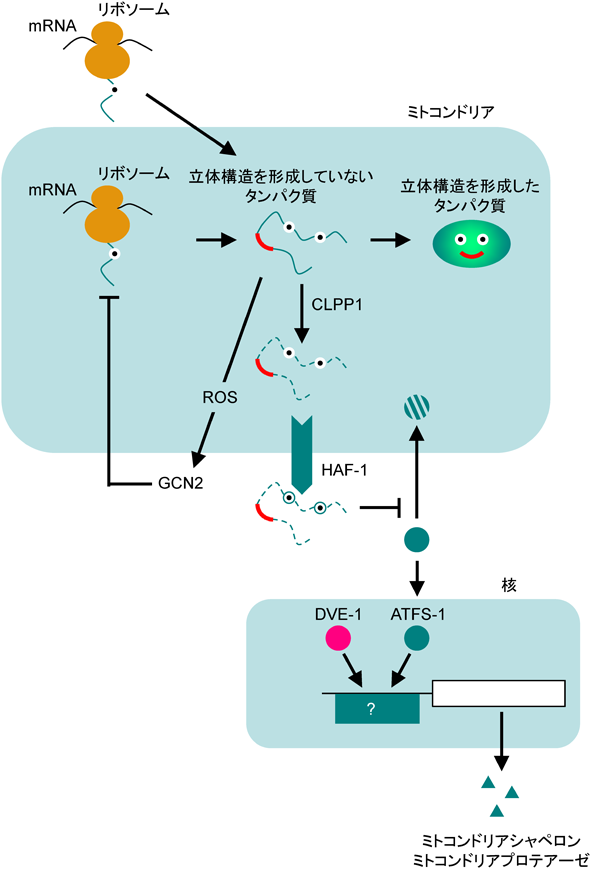
もう一つの経路であるROS-GCN2経路は,小胞体ストレス応答のPERK経路とよく似ている.ミトコンドリアに異常タンパク質が蓄積してミトコンドリアの機能が不全となると,ミトコンドリアに活性酸素(reactive oxygen species:ROS)が産生され,このROSによってキナーゼであるGCN2が活性化されてeIF2αをリン酸化し,翻訳を抑制することでミトコンドリアストレス状態がそれ以上悪化しないようにしている12).
線虫で発見されたATFS-1の機能的な哺乳類ホモログとして最近ATF5が報告され,ATFS-1経路が哺乳類でも保存されていることがわかった21).ATF5はアミノ酸飢餓やカドミウムなどのストレス応答や筋肉細胞の機能に関与しているという報告もあり,ミトコンドリアストレス応答とこれらのストレス応答の関係が注目を集めている22).ATF5とATFS-1はどちらもbasic leucine zipper型の転写因子であるが,アミノ酸上の相同性はホモログというほどには高くない.ATF5がATFS-1のホモログであるかどうかは,議論のあるところである.
ROS-GCN2経路も保存されていて,哺乳類の場合にはGCN2ではなくPKRがeIF2αをリン酸化する23).哺乳類にもGCN2があるのに,わざわざウイルス感染などによって活性化するPKRを用いている理由は不明である.
哺乳類には,あと三つ応答経路が知られている.一つは,CHOP経路である.ミトコンドリアマトリクス内に構造異常タンパク質が蓄積すると,MEK-JNK2-cJun経路が活性化して転写因子CHOPの発現を誘導し,CHOPがCHOP elementというエンハンサー(コンセンサス配列はGGT TGC A)に結合することによってミトコンドリアのシャペロンやプロテアーゼ遺伝子の転写を誘導する24).
もう一つの経路は,IMS-UPR経路である25, 26).ミトコンドリアの外膜と内膜の間の空間(膜間部分,intermembrane space:IMS)に構造異常タンパク質が蓄積するとROSが発生し,このROSがキナーゼAktの活性化を介してミトコンドリア膜に存在しているエストロゲン受容体αを活性化し,その結果転写因子NRF1を活性化する26).活性化したNRF1は呼吸鎖の諸因子の遺伝子発現を誘導し,ミトコンドリアの機能を増強しようとする.活性化したエストロゲン受容体はプロテアソームやミトコンドリアのプロテアーゼOMIを活性化し,膜間部分に蓄積した異常タンパク質の分解能力を高めている.
最後の経路は,SIRT3経路である27).この経路はミトコンドリアに存在する脱アセチル化酵素であるSIRT3が主たる制御因子であり,マイトファジーや抗酸化ストレス応答を制御しているが,その詳細は不明である.
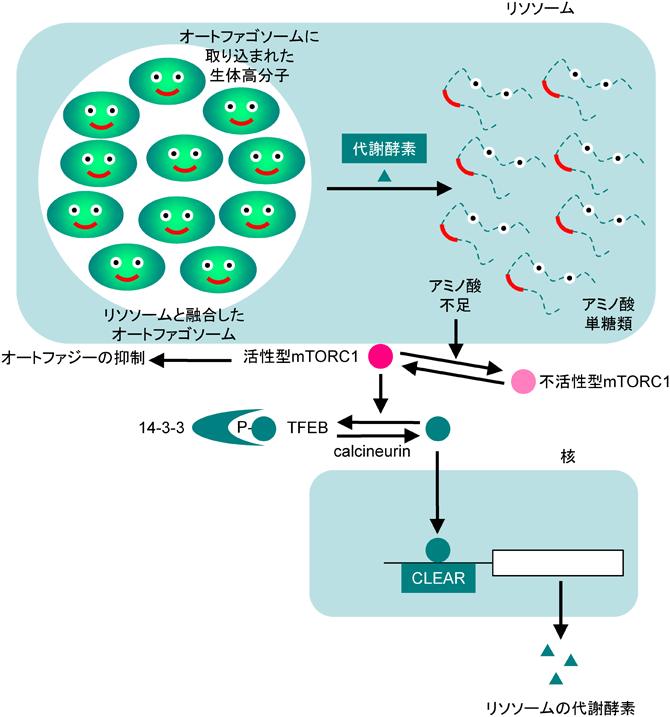
リソソームの量的調節機構であるlysosomal stress responseは,TFEBという転写因子が制御の中心である28–35)(図8).リソソームの主たる機能はタンパク質や多糖類などの生体高分子の分解であるが,これらの生体高分子化合物は主として飢餓時にオートファジーによってリソソームへと運ばれる(もちろん,飢餓時以外のオートファジーも存在するが,飢餓時のオートファジーは非選択的で,非常に強力である).したがって,飢餓時のオートファジーを起こす際にはリソソームの機能を大規模に亢進する必要がある.栄養(特にアミノ酸)が豊富に存在するときには,キナーゼ複合体mTORC1はv-ATPaseであるRagulator複合体によって細胞質ゾルからリソソーム膜上に移行し,低分子Gタンパク質Rhebによって活性化される.活性化したmTORC1はリボソームタンパク質であるS6Pや,翻訳開始因子eIF4Eの結合因子に結合する4E-BP1をリン酸化することによってタンパク質の翻訳を向上させるとともに,オートファジーの調節因子であるULK1をリン酸化することでオートファジーの開始を抑制する.また,mTORC1はTFEBをリン酸化し,リン酸化されたTFEBは14-3-3タンパク質と結合することで細胞質に不活性な状態で繋留される.アミノ酸が枯渇するとmTORC1はリソソーム膜から解離して不活性化し,その結果ULKが脱リン酸化されてオートファジーが開始するとともに,TFEBは脱リン酸化酵素であるcalcineurinによって脱リン酸化し,14-3-3から解離することで核へ移行してエンハンサーであるCLEAR配列に結合し,リソソーム機能やオートファジーに関与する遺伝子の転写を誘導する.このTFEB経路はリソソームに蓄積した高分子化合物を分解処理するために必須の機構であり,ヒトの疾患(Pompe病などのリソソーム蓄積症やフォールディング病であるハンチントン病など)と深い関係が報告されている.ただし,TFEBのホモログは酵母や線虫には存在しないことから,下等生物におけるリソソームストレス応答の分子機構は哺乳類のTFEB経路とは少し異なっているのかもしれない.
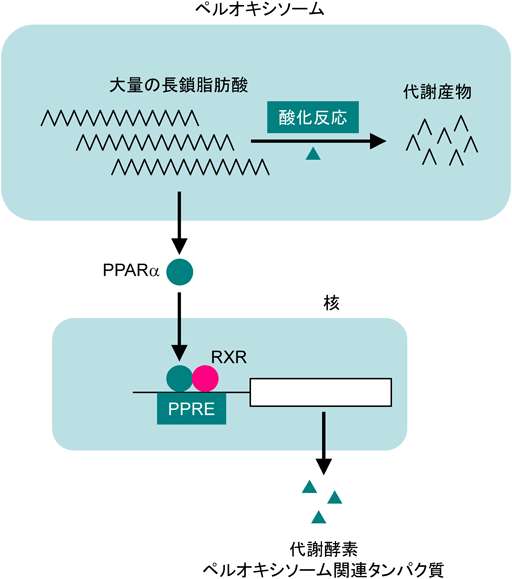
ペルオキシソームの重要な機能の一つは,長鎖脂肪酸を酸化反応によって分解することである.脂質を大量に摂取すると,細胞に蓄積した長鎖脂肪酸が転写因子PPARαに直接結合して活性化し,その結果ペルオキシソームの機能を担う遺伝子の転写が誘導されてペルオキシソームの機能が亢進する36)(図9).高脂血症などの治療薬であるフィブレートの作用点もこのPPARαであり,ペルオキシソームを増やして長鎖脂肪酸の代謝を亢進することで高脂血症を緩和する37).
前述したように,小胞体ストレス応答の研究は,もともとは小胞体の量的調節機構として研究が始められたのではなく,小胞体ストレス(立体構造が正常でないタンパク質が小胞体内腔に蓄積する状態)という恒常性破綻に対応する生体防御機構としてMary-Jane Gething博士(UT-South Western)によって研究が始められた.その後,Gething研のポスドクだった森和俊博士(現京都大学)と,Günter Brobel研から独立したPeter Walter博士(UCSF)が中心となって小胞体ストレス応答の分子機構を解明し,この研究分野を大きく発展させた.これらの業績により,森博士とWalter博士は2014年のラスカー賞を受賞した.
哺乳類の小胞体ストレス応答機構には,三つの応答経路が存在する4)(図10).それぞれの経路には,小胞体ストレスを感知するセンサーがあり,そのセンサーによって活性化される転写因子とその転写因子が結合するエンハンサー配列,そして最終的に転写が誘導される標的遺伝子がセットになって存在している.第一の経路であるATF6経路は小胞体の膜貫通タンパク質であるpATF6(P)がセンサー分子であり,小胞体ストレス(小胞体内腔に構造異常タンパク質が蓄積する状態)を感知すると小胞輸送によってゴルジ体へと運ばれ,そこで待ち構えているプロテアーゼS1PとS2Pによって切断を受け,細胞質側の転写因子部分pATF6(N)(Nはnuclear formを表す)が? 膜から遊離して核へ移行し,エンハンサーであるERSEに結合してBiPなどの小胞体シャペロンの転写を誘導する.
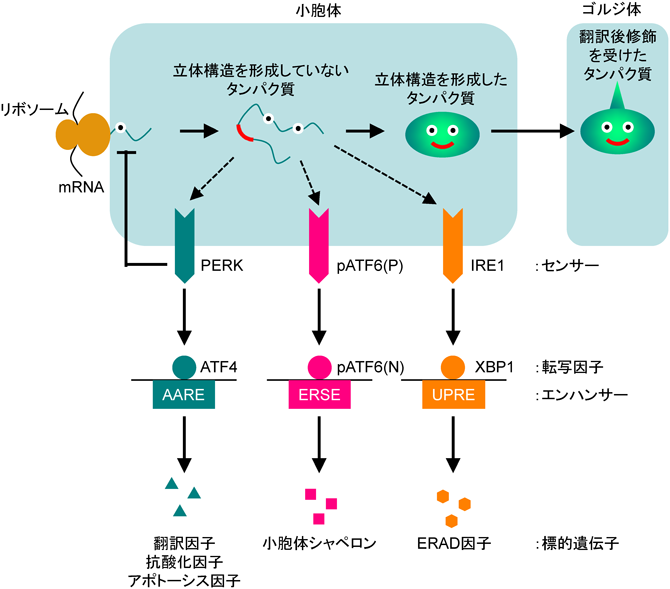
ATF6経路とIRE1経路,PERK経路が存在し,それぞれ独自のセンサーや転写因子,エンハンサー,標的遺伝子を持っている.経路間には複雑なクロストークがあるが,本図では省略する.
第二の経路であるIRE1経路のセンサーは,小胞体膜貫通型RNaseであるIRE1である.IRE1は小胞体ストレスを感知すると多量体化して活性化し,XBP1の前駆体mRNAをスプライシングして成熟型mRNAへと変換する.この成熟型mRNAから活性型転写因子pXBP1(S)(Sはspliced formを意味する)が翻訳され,エンハンサー配列UPREに結合して主として小胞体で生じた不良品タンパク質の分解機構(ER-associated degradation:ERAD)の因子の発現を誘導する.
第三の経路であるPERK経路のセンサーは,小胞体膜貫通型のキナーゼである.小胞体ストレスを感知して活性化したPERKは翻訳開始因子の一つであるeIF2のαサブユニット(eIF2α)をリン酸化し,翻訳を一時的に抑制することで,それ以上小胞体に構造異常タンパク質が蓄積しないようにしている.また,全体的な翻訳が低下することによって転写因子ATF4の翻訳は逆に上昇し,エンハンサーであるAAREを介して翻訳に関わる因子や抗酸化ストレスに対処する因子,アポトーシスを誘導する転写因子CHOPなどの転写を誘導する.
今のところ論文として報告されているゴルジ体ストレス応答経路は上記の3経路だけであるが,ゴルジ体は小胞体に比べて構造や機能が複雑であることから,応答経路もまだまだ未知のものがたくさんあると想像される.たとえば,ゴルジ体の重要な機能として翻訳後修飾があるが,糖鎖修飾だけでもN型やプロテオグリカン型,ムチン型などがあり,それぞれ別個の修飾酵素群を用いている(一部は流用されている).軟骨細胞ではプロテオグリカン型,粘液分泌細胞ではムチン型が多いので,これらの糖鎖修飾酵素群は別々の経路で制御されている可能性が高い.
また,小胞体やゴルジ体だけでなく他の細胞小器官の量的調節機構についても解析が進みつつある.このように,細胞小器官に量的調節機構が存在することはかなり確かなことと考えられるようになってきた38).今後さらに研究が進展し,細胞生物学の教科書に細胞小器官の量的調節機構の章が加わることを楽しみにしている.
以上,ゴルジ体ストレス応答を中心に細胞小器官の量的調節機構について解説した.蛇足であるが,小職の研究室の論文は,2009年以降特別の事情がない限りすべて日本細胞生物学会誌であるCell Structure and Function(大野博司編集長)に出すことにし,DORA宣言にも署名した.これは,(1)この雑誌がきわめてフェアで優れた雑誌であること,(2)「科学を出世の道具に使わない」というポリシーのためである.
1) Taniguchi, M., Nadanaka, S., Tanakura, S., Sawaguchi, S., Midori, S., Kawai, Y., Yamaguchi, S., Shimada, Y., Nakamura, Y., Matsumura, Y., Fujita, N., Araki, N., Yamamoto, M., Oku, M., Wakabayashi, S., Kitagawa, H., & Yoshida, H. (2015) Cell Struct. Funct., 40, 13–30.
2) Miyata, S., Mizuno, T., Koyama, Y., Katayama, T., & Tohyama, M. (2013) PLoS ONE, 8, e69732.
3) Reiling, J.H., Olive, A.J., Sanyal, S., Carette, J.E., Brummelkamp, T.R., Ploegh, H.L., Starnbach, M.N., & Sabatini, D.M. (2013) Nat. Cell Biol., 15, 1473–1485.
4) Yoshida, H. (2007) FEBS J., 274, 630–658.
5) Oku, M., Tanakura, S., Uemura, A., Sohda, M., Misumi, Y., Taniguchi, M., Wakabayashi, S., & Yoshida, H. (2011) Cell Struct. Funct., 36, 1–12.
6) Dinter, A. & Berger, E.G. (1998) Histochem. Cell Biol., 109, 571–590.
7) Nishihara, S. (2014) CMP-sialic acid transporter (CST), Solute carrier family 35 member A1 (SLC35A1). in Handbook of Glycosyltransferases and Related Genes (Taniguchi, N. ed.), pp. 1369–1377, Springer.
8) Xu, Y.X., Liu, L., Caffaro, C.E., & Hirschberg, C.B. (2010) J. Biol. Chem., 285, 24600–24608.
9) Sohda, M., Misumi, Y., Yamamoto, A., Yano, A., Nakamura, N., & Ikehara, Y. (2001) J. Biol. Chem., 276, 45298–45306.
10) Taniguchi, M., Sasaki-Osugi, K., Oku, M., Sawaguchi, S., Tanakura, S., Kawai, Y., Wakabayashi, S., & Yoshida, H. (2016) Cell Struct. Funct., 41, 93–104.
11) Lin, Y.F. & Haynes, C.M. (2016) Mol. Cell, 61, 677–682.
12) Baker, B.M., Nargund, A.M., Sun, T., & Haynes, C.M. (2012) PLoS Genet., 8, e1002760.
13) Haynes, C.M., Petrova, K., Benedetti, C., Yang, Y., & Ron, D. (2007) Dev. Cell, 13, 467–480.
14) Haynes, C.M., Yang, Y., Blais, S.P., Neubert, T.A., & Ron, D. (2010) Mol. Cell, 37, 529–540.
15) Nargund, A.M., Pellegrino, M.W., Fiorese, C.J., Baker, B.M., & Haynes, C.M. (2012) Science, 337, 587–590.
16) Nargund, A.M., Fiorese, C.J., Pellegrino, M.W., Deng, P., & Haynes, C.M. (2015) Mol. Cell, 58, 123–133.
17) Pellegrino, M.W. & Haynes, C.M. (2015) BMC Biol., 13, 22.
18) Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., Haynes, C.M., & Chen, D. (2015) Science, 347, 1374–1377.
19) Tian, Y., Garcia, G., Bian, Q., Steffen, K.K., Joe, L., Wolff, S., Meyer, B.J., & Dillin, A. (2016) Cell, 165, 1197–1208.
20) Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S.D., Quiros, P.M., Steffen, K.K., Williams, E.G., Mouchiroud, L., Tronnes, S.U., Murillo, V., Wolff, S.C., Shaw, R.J., Auwerx, J., & Dillin, A. (2016) Cell, 165, 1209–1223.
21) Pellegrino, M.W., Nargund, A.M., Kirienko, N.V., Gillis, R., Fiorese, C.J., & Haynes, C.M. (2014) Nature, 516, 414–417.
22) Abe, T., Kojima, M., Akanuma, S., Iwashita, H., Yamazaki, T., Okuyama, R., Ichikawa, K., Umemura, M., Nakano, H., Takahashi, S., & Takahashi, Y. (2014) J. Biol. Chem., 289, 3888–3900.
23) Rath, E., Berger, E., Messlik, A., Nunes, T., Liu, B., Kim, S.C., Hoogenraad, N., Sans, M., Sartor, R.B., & Haller, D. (2012) Gut, 61, 1269–1278.
24) Ryan, M.T. & Hoogenraad, N.J. (2007) Annu. Rev. Biochem., 76, 701–722.
25) Radke, S., Chander, H., Schafer, P., Meiss, G., Kruger, R., Schulz, J.B., & Germain, D. (2008) J. Biol. Chem., 283, 12681–12685.
26) Papa, L. & Germain, D. (2011) J. Cell Sci., 124, 1396–1402.
27) Papa, L. & Germain, D. (2014) Mol. Cell. Biol., 34, 699–710.
28) Martina, J.A., Chen, Y., Gucek, M., & Puertollano, R. (2012) Autophagy, 8, 1–12.
29) Pena-Llopis, S., Vega-Rubin-de-Celis, S., Schwartz, J.C., Wolff, N.C., Tran, T.A., Zou, L., Xie, X.J., Corey, D.R., & Brugarolas, J. (2011) EMBO J., 30, 3242–3258.
30) Roczniak-Ferguson, A., Petit, C.S., Froehlich, F., Qian, S., Ky, J., Angarola, B., Walther, T.C., & Ferguson, S.M. (2012) Sci. Signal., 5, ra42.
31) Settembre, C., De Cegli, R., Mansueto, G., Saha, P.K., Vetrini, F., Visvikis, O., Huynh, T., Carissimo, A., Palmer, D., Jurgen Klisch, T., Wollenberg, A.C., Di Bernardo, D., Chan, L., Irazoqui, J.E., & Ballabio, A. (2013) Nat. Cell Biol., 15, 647–658.
32) Settembre, C., Di Malta, C., Polito, V.A., Garcia Arencibia, M., Vetrini, F., Erdin, S., Erdin, S.U., Huynh, T., Medina, D., Colella, P., Sardiello, M., Rubinsztein, D.C., & Ballabio, A. (2011) Science, 332, 1429–1433.
33) Settembre, C., Zoncu, R., Medina, D.L., Vetrini, F., Erdin, S., Huynh, T., Ferron, M., Karsenty, G., Vellard, M.C., Facchinetti, V., Sabatini, D.M., & Ballabio, A. (2012) EMBO J., 31, 1095–1108.
34) Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J.A., Annunziata, F., Zare, H., Polishchuk, R., Puertollano, R., Parenti, G., Ballabio, A., & Raben, N. (2013) EMBO Mol. Med., 5, 691–706.
35) Sardiello, M., Palmieri, M., di Ronza, A., Medina, D.L., Valenza, M., Gennarino, V.A., Di Malta, C., Donaudy, F., Embrione, V., Polishchuk, R.S., Banfi, S., Parenti, G., Cattaneo, E., & Ballabio, A. (2009) Science, 325, 473–477.
36) Chakravarthy, M.V., Lodhi, I.J., Yin, L., Malapaka, R.R., Xu, H.E., Turk, J., & Semenkovich, C.F. (2009) Cell, 138, 476–488.
37) Green, S. (1995) Mutat. Res., 333, 101–109.
38) Sasaki, K. & Yoshida, H. (2015) J. Biochem., 157, 185–195.

兵庫県立大学教授(大学院生命理学研究科).博士(理学).
1964年大阪府吹田市に生まる.87年京都大学理学部卒業.89年同大学院理学研究科修士課程修了.94年同大学院理学研究科博士課程修了.博士研究員(HSP研究所,メルボルン大学,京都大学,JSTさきがけ)を経て,2004年京都大学大学院理学研究科准教授.10年より現職.
研究テーマと抱負細胞小器官の量的調節機構(特に,ゴルジ体ストレス応答).
ウェブサイトhttps://sites.google.com/site/hiderouoshidalab2/home
趣味自動二輪

This page was created on 2017-02-06T13:22:18.598+09:00
This page was last modified on 2017-04-17T09:28:53.210+09:00
このサイトは(株)国際文献社によって運用されています。