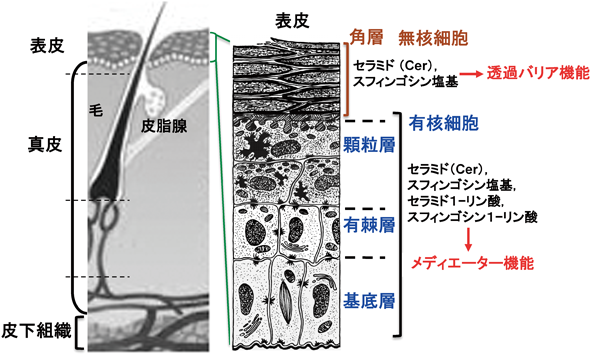
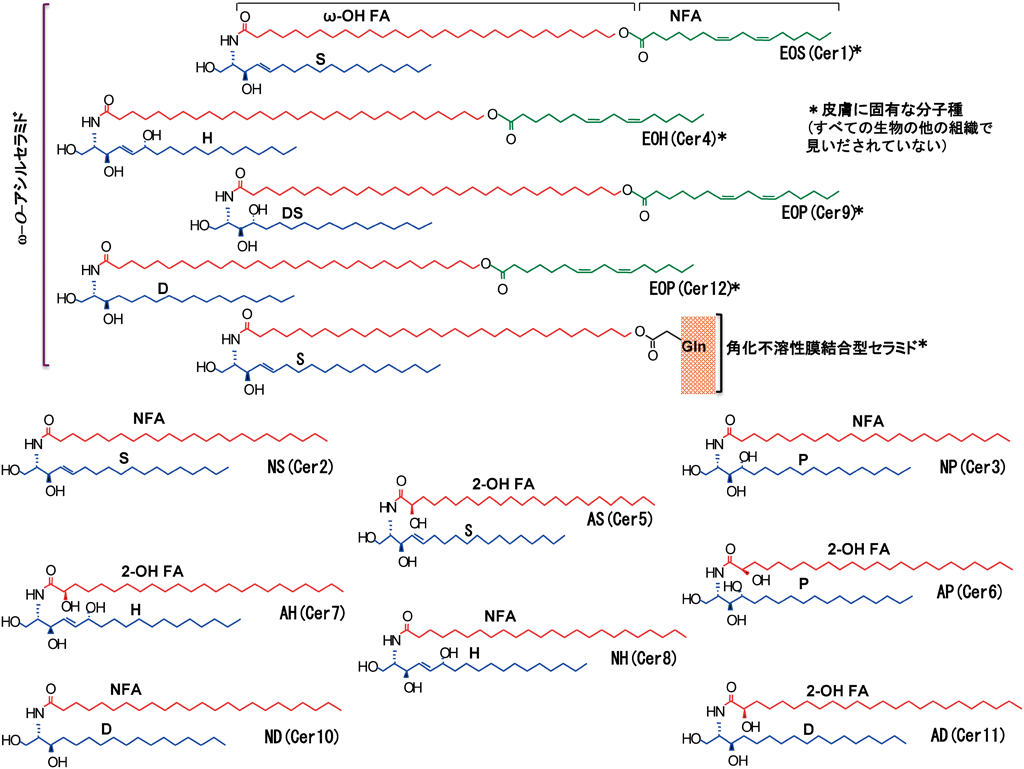
分化後期(顆粒層)のケラチノサイトは12分子種のセラミドを合成する(図3,Cer1~12).これらのセラミド分子の多様性は,セラミドを構成する脂肪酸とスフィンゴシン塩基のヒドロキシ化(非ヒドロキシ,2-ヒドロキシおよび末端のω-ヒドロキシ)の多様性に起因する.ヒドロキシ化の多様性に加えて,脂肪酸とスフィンゴシン塩基の鎖長にも多様性がある.このような多様な構造のセラミドが,コレステロールと脂肪酸とともに角層の安定な脂質多層膜(ラメラ)構造の形成に寄与している(図2)6, 7).
表皮顆粒層で合成されたセラミドのほとんどは,グルコシルセラミド合成酵素によりグルコシルセラミドへ,また,スフィンゴミエリン合成酵素によりスフィンゴミエリンへ代謝され,それらの一部は分化後期のケラチノサイトに含まれる層板顆粒(ラメラ顆粒,ラメラボディ)に輸送され蓄積する.表皮以外には肺にも層板顆粒が含まれている.両者とも形態的に類似するが,肺の層板顆粒は肺サーファクタント(ホスファチジルコリン80~90%と,ホスファチジルグリセロール5~10%)を貯留しており,含有物にケラチノサイトと相違を示す8).層板顆粒の内容物は有核ケラチノサイトの細胞内で分解されたり,細胞内に放出されたりしないと考えられている.したがって,層板顆粒は角層に必要な成分を格納し,角層に届ける細胞内小器官と換言できる.層板顆粒から角質細胞間に放出されたグルコシルセラミド,スフィンゴミエリンは,酸性型のβ-グルコセレブロシダーゼとスフィンゴミエリナーゼによりそれぞれ加水分解を受け,セラミドに変換される.生成したセラミドは肪酸やコレステロールとラメラ構造体を作り,表皮透過バリアを形成する.ラメラ構造体には,これら主要な脂質成分以外にも少量成分ながらセラミド代謝産物のスフィンゴシンとジヒドロスフィンゴシン(スフィンガニン)が含まれ,ラメラ構造の安定化と充填性の高い表皮透過バリアの形成に寄与している9).角層に含まれる計12分子種のセラミドはすべてグルコシルセラミドから産生されるが,スフィンゴミエリンからは2種の産生にとどまる(図3のセラミド分子種NS, AS10, 11).遺伝性のスフィンゴ糖脂質代謝異常症のゴーシェ病のうち酵素活性がない,あるいは残存酵素活性がきわめて低いゴーシェ病2型においては,グルコシルセラミドから角層のセラミドの生成ができず,結果として皮膚の透過バリア形成不全で死に至る12).一方,酸性型のスフィンゴミエリナーゼ活性が低下するニーマン・ピック病A型とB型においては,急性期的な透過バリア崩壊からの回復性は低下する13)ものの患者に皮膚症状は報告されていない.この事実はグルコシルセラミド由来のセラミドがバリア形成に欠かせないことを示唆している.
12種のセラミド分子種の中で,3種のセラミド(ω-O-アシルセラミド)は,分化した陸上哺乳動物のケラチノサイトでのみ合成される.12種に加えてω-O-アシル化されていないω-ヒドロキシセラミドは,ω位の水酸基が角化不溶性膜の細胞間隙側に位置するグルタミン酸/グルタミンに富むペプチドに共有結合し,角質細胞脂質外膜(corneocyte lipid-bound envelope:CLE)を形成している.このタンパク質に結合したセラミドは結合型セラミド,それ以外のセラミドは非結合型セラミドと呼称されている14).
ω-O-アシルセラミドは,極長鎖脂肪酸(炭素鎖長28~34)の合成,その末端ω位が水酸基化,極長鎖脂肪酸のスフィンゴシン塩基への結合(セラミド合成),およびω位水酸基のアシル化(ほとんどがリノール酸)の4段階を経て生合成される.最近,これらのすべての生合成過程が明らかとなった.筆者らは,その最初のステップである極長鎖脂肪酸合成が脂肪酸鎖長延長酵素(ELOVL4)によることを明らかにした15, 16).その後,ELOVL4の基質となる長鎖脂肪酸はELOVL1により合成されること16)が明らかとなり,次いで,残りの3ステップが解き明かされていった.第2ステップではCYP4F22により極長鎖脂肪酸のω位ヒドロキシ化が起き17, 18),第3ステップでセラミド合成酵素(Cer3)によりω-ヒドロキシセラミドが生成され,最後の第4ステップでω-O-アシル化され,ω-O-アシルセラミドが生成される.この第4ステップの酵素はいまだ同定されていないが,筆者らはドルフマン・シャナリン魚鱗癬患者の角層脂質の分析から以下のような興味ある知見を得ている.患者の角層ではトリグリセリドが蓄積し,ω-O-アシルセラミドおよび結合型ω-ヒドロキシセラミド量が低下していた19).さらにイメージング質量分析により,表皮ω-O-アシルセラミドの低下とリノール酸を含むトリグリセリドの蓄積を確認した20).ドルフマン・シャナリン魚鱗癬はCGI-58(comparative gene identification-58,脂肪細胞トリグリセリドリパーゼの活性化因子)の遺伝子変異が原因となる疾患である.患者角層の脂質異常から,ω-ヒドロキシセラミドのω位水酸基のアシル化に必要なリノール酸は,CGI-58により活性化されるトリグリセリドリパーゼによりトリグリセリドから供給されることが明らかとなった.CGI-58は脂肪細胞トリグリセリドリパーゼ(adipose triglyceride lipase:ATGL)の活性化タンパク質として同定されたが,ATGL欠損マウスにおいては表皮透過バリア機能に異常が観察されない21).これに対して,ドルフマン・シャナリン魚鱗癬,あるいは,CGI-58欠損マウスは,ω-O-アシルセラミドの低下と表皮透過バリア形成不全を起こす.したがって,表皮にはCGI-58で活性化される別のトリグリセリドリパーゼが発現していると考えられる.ω-O-アシルセラミドのリノール酸残基は,12R-リポキシゲナーゼと表皮型リポキシゲナーゼ-3およびエポキシドの分解により,順次,リノール酸9-ヒドロキシエポキシド,リノール酸エポキシアルコール,さらに,トリヒドロキシリノール酸となって,セラミドのω水酸基は角化不溶性膜に結合する(結合型セラミド)22).
一方,ヒドロキシセラミドのかなりの部分を占めるα-ヒドロキシセラミドのα-ヒドロキシ脂肪酸(2位水酸化脂肪酸)は,2位水酸化酵素(FA 2-hydroxylase:FA2H)により生成される23).筆者らは,ケラチノサイトにFA2Hが発現することを示し,さらに,その遺伝子の発現を低下させた培養ヒトケラチノサイトでは2-ヒドロキシ脂肪酸と2-ヒドロキシ脂肪酸含有セラミドが低下すること,および,分化と物質透過バリア形成の異常が起きることを見いだした24).この事実は,これら脂質とFA2Hが,ケラチノサイトの機能に重要であることを示唆する.しかし,遺伝性痙性対麻痺タイプ35[hereditary spastic paraplegia(SPG35)]の病因となるFA2Hの変異した患者に皮膚症状は報告されていない25, 26).したがって,表皮においてはFA2H以外の脂肪酸2位水酸化酵素が発現している可能性が高い.
以上のように,いまだ同定されていないω-O-アシル化酵素,トリグリセリドリパーゼ,および,脂肪酸2位水酸化酵素を除けば,表皮で生合成される12種の多様な表皮セラミドの全生合成過程がほぼ明らかとなっている.一方,表皮を含めて,セラミドの代謝調節機構は,以下に示すような若干の検討がなされているにすぎない.
核受容体のペルオキシソーム増殖剤応答性受容体(PPAR)α, PPARβ/δおよび肝臓核受容体(LXR)は,表皮のスフィンゴ脂質を含めた脂質の合成に影響している.ヒトの培養ケラチノサイトを用いた検討から,PPARαはセリンパルミトイルトランスフェラーゼ,アシルCoA合成・酸化酵素とHMG–CoA合成酵素2の発現を高めること27, 28),PPARβ/δリガンドを塗布したマウスの表皮で表皮透過バリアを形成するセラミドの合成が高まること29),また,PPARα, PPARβ/δおよびLXRリガンド塗布により,β-グルコセレブロシダーゼとスフィンゴミエリナーゼの遺伝子発現が亢進することなどが報告されている.核受容体であるビタミンD受容体(VDR)の転写共役因子VDR-interacting protein(DRIP)はケラチノサイトの分化の初期に発現が高まるのに対して,steroid receptor coactivator(SRC)2と3は,分化後期に発現が高まる.分化初期にはDRIPが,後期にはSRC2と3が,ビタミンD受容体の活性化を介して,違ったタンパク質や脂質合成を調節している.筆者らは,ビタミンD受容体とこれらの転写共役因子がセラミドとグルコシルセラミドの産生調節に関与するが,スフィンゴミエリンの産生調節には関わらないことを明らかにした30).
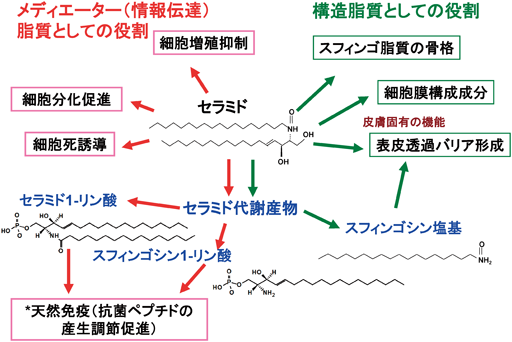
7. 皮膚におけるセラミドとセラミド代謝産物のメディエーターとしての役割
1)セラミド
セラミドが赤芽球分裂促進作用を示すことが,1974年にR.B. Clayton博士(スタンフォード大学)らにより55)報告されている.炭素鎖長24のセラミドに最も高い活性が認められた55).しかし,その後は,セラミド自身のメディエーターとしての役割研究は進まなかった.
1986年のYusuf A. Hannun博士(現ストニーブルック大学)とRobert M. Bell博士(デューク大学)によるスフィンゴシンによるプロテインキナーゼC阻害作用の発見に続き,同研究者らとBell研に留学していた岡崎俊郎博士(現金沢医科大学)56–58)によるセラミドのヒト白血病細胞の分化誘導作用の解明がセラミド研究の転換を導いた.ほぼ同時期に,これら研究者とは別にRichard Kolesnick博士(スロンケタリング記念がんセンター)は,スフィンゴミエリナーゼの活性化により,増加したセラミドがヒト白血病細胞の分化誘導に作用することを報告している59).これら4科学者が,セラミドのメディエーターとしての研究発展の礎を作ったといっても過言でない.難容性のセラミドの生理活性を調べることは困難であったが,彼らは,細菌由来のスフィンゴミエリナーゼと合成した短鎖のセラミド(酸アミド結合脂肪酸鎖2~8)を利用し,この難点を克服した.しかし,生体内に短鎖のセラミドは含まれていないことから,この実験手法は人為的すぎるとの見解もあった.その後,短鎖のセラミドは細胞内に取り込まれた後,セラミダーゼによりスフィンゴシン塩基と短鎖脂肪酸に分解され,次いで,生成されたスフィンゴシン塩基と細胞内にある長鎖脂肪酸からセラミド合成酵素により長鎖セラミドが合成されることが明らかになり60),人為的作用の懸念は減った.細胞内で長鎖型のセラミドが増加していることを確認すれば,細胞内のセラミドやスフィンゴシン1-リン酸量を高める容易な実験手法として利用できる.
皮膚においてもセラミドのメディエーターとしての役割が検討されている.外から添加した短鎖セラミドは培養ケラチノサイトの分化を促進する61).筆者らは,B波長の紫外線照射や酸化ストレスで産生の高まったセラミドがアポトーシスを誘導することを明らかにした62).このことは,セラミドが生理的にもメディエーターとして役割を果たすことを示唆する.この検討の中で,低線量の紫外線はアポトーシスを誘導しないが,細胞死を起こす高線量と同程度線量を照射すると,照射後の初期(6~8時間)に細胞内のセラミド量が高まる63).その後,低線量照射の場合,セラミド量が低下していくのに対して,高線量の照射の場合,24時間後も細胞内のセラミド量が高まっており,アポトーシスを起こした細胞数も増加した63).セラミドにより誘導されるアポトーシスは制がん剤や放射線療法の作用機作の一つとなる.一方,セラミダーゼ,グルコシルセラミド合成酵素,スフィンゴミエリン合成酵素,および,スフィンゴシンキナーゼの恒常的な活性化により,がん細胞は制がん剤や放射療法耐性となる64).そこで,治療の効果を維持するため,制がん剤や放射線療法とスフィンゴ脂質代謝阻害剤の併用治療も検討されている64, 65).筆者らの得た知見は,薬剤耐性がんを産むセラミドの代謝機構が,正常なケラチノサイトでは紫外線や酸化ストレスにより誘導されるセラミド依存的なアポトーシス回避機構としても働くことを示唆している63).ゴルジ体で合成されるスフィンゴミエリンの基質となるセラミドは,小胞体からセラミド移送タンパク質(CERT)により移送されるが,筆者らは,紫外線や酸化ストレスでCERTが安定な三量体を形成しセラミド移送機能が低下することを見いだした66).したがって,セラミドのスフィンゴミエリンへの代謝機構は,ストレス下で十分に機能しない可能性が高い.ケラチノサイトを含めて,哺乳動物において,セラミドは,酵素の活性化(セラミド活性化セリン・トレオニンホスファターゼ,プロテインホスファターゼ1Aと2A,プロテインキナーゼCζ他)67)を介して細胞機能に影響を与える.このような機構に加え,セラミドは物理的にミトコンドリア膜に小孔を作り,アポトーシスを誘導する67).また,セラミドは膜の曲率を変える物性を持つことから,細胞膜の構造変化を介して細胞に影響を与えている可能性が高い.
2)スフィンゴシン塩基
スフィンゴシンを外から添加することにより,培養ケラチノサイトの増殖が抑制される61).また,4-ヒドロキシジヒドロスフィンゴシン(フィトスフィンゴシン)は培養ケラチノサイトの分化を誘導する68).マウスの皮膚を対象とした検討から,ホルボールエステル塗布で誘導される炎症性の皮膚肥厚は,フィトスフィンゴシン塗布で軽減される68).しかし,これらの報告において,スフィゴシンやフィトスフィンゴシンが,各々スフィンゴシン1-リン酸とフィトスフィンゴシン1-リン酸に代謝あるいはセラミドに再合成され,増殖抑制,分化促進あるいは抗炎症作用を示したかどうかは明らかにされていない.
3)スフィンゴシン1-リン酸
スフィンゴシン1-リン酸が細胞で生合成されることは1970年に明らかになっていたが69),その生理学的な役割は検討されていなかった.1990年の初頭にSarah Spiegel博士(バージニア・コモンウェルス大学)ら,ならびに五十嵐靖之博士(北海道大学)らにより,スフィンゴシン1-リン酸がメディエーター機能を持つことが明らかにされた.その後,細胞膜に発現するスフィンゴシン1-リン酸受容体も同定されたことから,スフィンゴシン1-リン酸の研究が急速に進んできた.スフィンゴシン1-リン酸は,細胞の種類,細胞が受ける刺激依存的にさまざまな作用を示す.皮膚において,スフィンゴシン1-リン酸は皮膚線維芽細胞,メラノーマ,脂肪細胞などの増殖を高めるが,ケラチノサイトでは増殖抑制,分化促進作用70–72)を示す.ケラチノサイトにおいて,スフィンゴシン1-リン酸は,スフィンゴシン1-リン酸受容体2(S1P2)への結合により,プロテインキナーゼCδを活性化する.その結果,Aktの脱リン酸化を引き起こし,インスリン受容体の活性化を介する増殖促進作用が阻害され,分化が誘導される73).ケラチノサイトの過増殖を伴う炎症性角化性皮膚疾患である乾癬では活性型ビタミンDが治療薬として用いられる.その作用機序の一つは,スフィンゴシン1-リン酸の産生を上昇させることによるケラチノサイトの増殖抑制と分化促進作用である74).
リンパ球は骨髄や胸腺と血流を介してリンパ節と脾臓間で循環する.スフィンゴシン1-リン酸濃度が高い場合,リンパ球のスフィンゴシン1-リン酸受容体の発現は低下し,リンパ球は活性化しない.血漿に比べスフィンゴシン1-リン酸濃度が低いリンパ節などではリンパ球のスフィンゴシン1-リン酸の受容体発現が高まり,リンパ球はスフィンゴシン1-リン酸によって活性化されて遊走性が高まり,リンパ節から血流に移動する75).アレルギー性物質による感作に先立ちスフィンゴシン1-リン酸を事前塗布するとアレルギー性接触皮膚炎が抑制される76).これは,スフィンゴシン1-リン酸により,アレルギー性接触皮膚炎成立に関わるランゲルハンス細胞(免疫担当樹状細胞)の皮膚からリンパ組織への遊走が抑制され,免疫反応の成立が低下したことによる.ヒトとイヌのアトピー性皮膚炎の皮膚においてはスフィンゴシン1-リン酸を分解するスフィンゴシン-1-リン酸リアーゼの遺伝子発現の亢進が認められ77–79),また,イヌのアトピー性皮膚炎において皮膚内のスフィンゴシン1-リン酸量の低下が確認されている78).スフィンゴシン1-リン酸の低下が過剰免疫反応を誘導し,アトピー性皮膚炎の発症と進行に関係していると考えられる.
4)セラミド1-リン酸
1990年を前後してセラミドの1位のリン酸化物,セラミド1-リン酸が哺乳動物細胞で生成されることが明らかになった80, 81).1990年代後半になり,Antonio Gomzez-Munoz博士(バスク大学)らにより,メディエーター機能が明らかにされていった.セラミド1-リン酸の受容体の存在が示唆され,その受容体の活性化を介した生理活性機構も報告82)されているが,受容体そのものは同定されていない.一方,Chales E. Chalfant博士(バージニア・コモンウェルス大学)らにより,セラミド1-リン酸が細胞内ホスホリパーゼA2への結合で同酵素を活性化し,産生されるアラキドン酸代謝産物のPGE2を介して,細胞機能の変化を与えることが明らかとなった83, 84).セラミド1-リン酸のホスホリパーゼA2への3次元的結合部位も同定されている85).小胞体からCERTにより移送されるセラミドが基質となり,セラミド1-リン酸が産生される.セラミド1-リン酸はセラミド1-リン酸輸送タンパク質により,細胞内を移送され,ホスホリパーゼA2の活性化を調節していると考えられている.異なった刺激に応答して,セラミド1-リン酸もスフィンゴシン1-リン酸と同様に,アポトーシス促進・抑制,増殖促進・抑制など多様な作用を示す86).
1)抗菌ペプチド
抗菌ペプチドは進化を超えて細菌,植物および動物で産生され,自然免疫の重要な因子として微生物感染から宿主細胞を防御している.哺乳動物では,上皮系細胞,リンパ球,好中球,単球やマクロファージなどで抗菌ペプチドが産生されている.抗菌ペプチドは電荷に富むペプチド(陽イオン性と陰イオン性の二つの型がある)で生体成分と結合しやすいため,細胞膜に結合あるいは融合し,膜に小孔を作って細胞内成分の漏出と細胞外成分の侵入により細胞機能を低下させる87, 88).ある種の抗菌ペプチドは細胞膜に融合または結合後に細胞内に取り込まれ,宿主の核酸やタンパク質に結合して代謝機能を不全にする88).したがって,抗菌ペプチドは菌株(ある種の真菌・ウイルスにも作用を示す)を問わない広域の抗菌スペクトルを示す.抗菌ペプチドを分解する酵素89)や抗菌ペプチドの排出ポンプの発現が誘導された場合には耐性菌が発生する90).しかし,抗生物質のように特定の代謝過程を阻害しないため抗菌ペプチド耐性菌の発生頻度は低い.
カテルシジン抗菌ペプチド(cathelicidin antimicrobial peptide)とβ-ディフェンシン1, 2および3は,表皮や皮脂腺の主要な抗菌ペプチドとして皮膚における微生物の増殖と浸潤を防いでいる.これらペプチドは常時産生されているが,細菌感染などにより産生が誘導される.
2)カテルシジンの産生調節機構
カテルシジンは,抗菌作用以外に細胞の増殖と遊走性を促進する作用を持つ多機能ペプチドである91).カテルシディンの過度な産生がある種の炎症やがん病変部で確認されており92),炎症惹起やがんの進行にも関わると考えられている.カテルシディンの転写は,プロモーター上にあるビタミンD受容体タンパク質の活性化により高まる.トール様受容体(TLR)2の活性化によりビタミンD受容体の活性化因子の産生が高まり,カテルシディンの産生が促進される93).また,リポ多糖によるTLR2/4受容体の活性化は活性型ビタミンD(1,25-ジヒドロキシビタモンD)の合成に必要なビタミンD-1-水酸化酵素の産生を高め,活性型ビタミンD量の増加を介してカテルシディン産生を増加させる94).しかし,マウスのカテルシディンのプロモーター上にはビタミンD受容体タンパク質結合配列が存在しない.また.ビタミンD欠乏者は,血中カテルシディン濃度が低く,結核の羅患率も高くなっていると報告95)されているが,ビタミンD欠乏者にあっても感染症の種類や病巣部位により相反する結果も出ている96).したがって,ビタミンD受容体非依存的なカテルシディン産生調節機構も存在すると考えるのが妥当である.以下に示すスフィンゴシン1-リン酸依存的機序は,ビタミンD受容体非依存的なカテルシディン転写調節機構の一つである97).
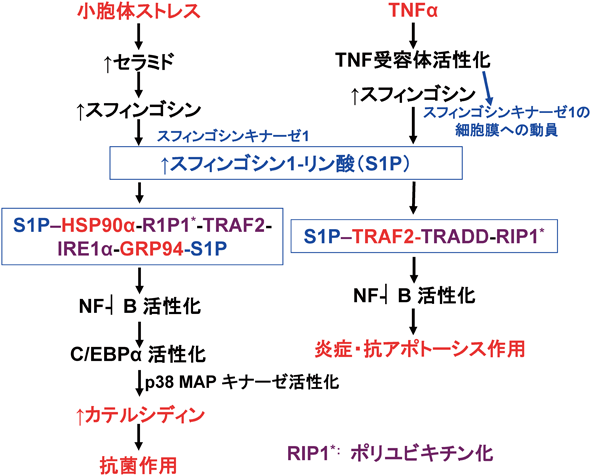
筆者らは,紫外線,酸化ストレスや表皮透過バリアの崩壊などの外来の侵襲が,細菌感染の痕跡なしに,カテルシディンの産生を高める事実に注目した.その機構を検討した結果,これら侵襲は小胞体(ER)ストレスを誘導し,転写調節因子のNF-κB,次いでc/EBPαを活性化し,カテルシディンの産生を高めることが明らかとなった97).小胞体ストレスは,セラミドの産生を高めること98)から,カテルシディンの産生亢進におけるセラミドのメディエーターとしての役割を調べた.小胞体ストレスにより産生の高まったセラミド,スフィンゴシンとスフィンゴシン1-リン酸のうち,スフィンゴシン1-リン酸がNF-κBを活性化してカテルシディンの産生亢進をもたらした99).スフィンゴシン1-リン酸は細胞膜に発現するGタンパク質共役受容体のスフィンゴシン1-リン酸受容体に結合し,その活性化を介して細胞機能に影響を与える.哺乳動物で5種の受容体が明らかにされている.このスフィンゴシン1-リン酸受容体経路以外に受容体非依存的な細胞内情報伝達経路も報告されているが,受容体依存的経路に比べて解明が進んでいない.スフィンゴシン1-リン酸受容体1, 2, 3,および,1と3のダブルノックアウトマウスの皮膚,さらに,スフィンゴシン1-リン酸受容体4と5の発現をsiRNAで抑制,あるいは,これら受容体の阻害剤と活性化剤を作用させた培養ケラチノサイトにおいても小胞体ストレスはカテルシディンの産生を高めた100).したがって,小胞体ストレスはスフィンゴシン1-リン酸受容体非依存的経路でNF-κBを活性化し,カテルシディンの産生を高めていると考えられた.
スフィンゴシン1-リン酸受容体非依存的なNF-κBの活性化として,TNFα受容体の活性化によりスフィンゴシン1-リン酸がTRAF2タンパク質に結合して情報伝達複合体を形成し,NF-κBの活性化を介して抗アポトーシス作用を示すことが明らかとなっている101).しかし,小胞体ストレスを介した場合,スフィンゴシン1-リン酸はTRAF2に結合せず,二つの熱ショックタンパク質(HSP90αと小胞体に在住するGRP94)と結合し,IRE1α(小胞体ストレスで活性化する小胞体タンパク質),TRAF2, RIP1とともに小胞体膜で情報伝達複合体を形成し,RIP1のポリユビキチン化を介して,転写調節因子NF-κBを活性化した100).TNFα受容体の活性化の場合,スフィンゴシン1-リン酸はHSP90αとGRP94に結合せず,TRAF2に結合した(図4)100).
スフィンゴシン1-リン酸は,アポトーシス誘導,抗アポトーシス,炎症抑制あるいは炎症惹起など,相反する作用を含めて多様な作用を示す.スフィンゴシン1-リン酸は,違った刺激に応じて,異なったタンパク質(スフィンゴシン1-リン酸受容体,TRAF2102),HSP90100),IRF1103)など)に結合することでさまざまな細胞反応の調節が可能となると考えられる.
3)β-ディフェンシン2および3
カテルシディン以外に,表皮透過バリアの崩壊によりβ-ディフェンシンの産生が高まる.筆者らは,カテルシディンと同様に,β-ディフェンシン2と3の産生が小胞体ストレスにより誘導されることを見いだした104).スフィンゴシン1-リン酸と同様に小胞体ストレスで産生の高まったセラミド1-リン酸はカテルシディンの産生には影響を与えないが,β-ディフェンシン2と3の合成を高めた.一方,スフィンゴシン1-リン酸はこれらβ-ディフェンシンの産生に影響しなかった.セラミド1-リン酸の増加により,ホスホリパーゼA2が活性化され産生の高まったプロスタグランジンのうちプロスタグランジンJ2は,PPARαおよびPPARβ/δを活性化した.次いで,これらPPARは,Srcタンパク質リン酸化酵素の活性化を介して転写調節因子STAT1とSTAT3を活性化し,β-ディフェンシン2と3の転写を高めた104)(図5).β-ディフェンシン2と3合成は,STAT1と3以外にNF-κBにより調節されていることが知られている105).しかし,小胞体ストレスによるセラミド1-リン酸を介する場合,NF-κBの活性化を抑制してもβ-ディフェンシン2と3の合成は影響を受けなかった.このことは,同一遺伝子の発現であっても上流の刺激が最終的な転写因子を決定することを示唆している.
引用文献References
1) Eckhart, L., Declercq, W., Ban, J., Rendl, M., Lengauer, B., Mayer, C., Lippens, S., Vandenabeele, P., & Tschachler, E. (2000) J. Invest. Dermatol., 115, 1148–1151.
2) Uchida, Y. & Park, K. (2016) Stratum. Corneum., Springer.
3) Oesch, F., Fabian, E., Oesch-Bartlomowicz, B., Werner, C., & Landsiedel, R. (2007) Drug Metab. Rev., 39, 659–698.
4) Scharschmidt, T.C., Vasquez, K.S., Truong, H.A., Gearty, S.V., Pauli, M.L., Nosbaum, A., Gratz, I.K., Otto, M., Moon, J.J., Liese, J., Abbas, A.K., Fischbach, M.A., & Rosenblum, M.D. (2015) Immunity, 43, 1011–1021.
5) Rice, R.H. & Green, H. (1977) Cell, 11, 417–422.
6) Mojumdar, E.H., Gooris, G.S., Groen, D., Barlow, D.J., Lawrence, M.J., Demé, B., & Bouwstra, J.A. (2016) Biochim. Biophys. Acta, 1858, 1926–1934.
7) Bouwstra, J., Gooris, G., & Ponec, M. (2002) J. Biol. Phys., 28, 211–223.
8) Schmitz, G. & Müller, G. (1991) J. Lipid Res., 32, 1539–1570.
9) Loiseau, N., Obata, Y., Moradian, S., Sano, H., Yoshino, S., Aburai, K., Takayama, K., Sakamoto, K., Holleran, W.M., Elias, P.M., & Uchida, Y. (2013) J. Dermatol. Sci., 72, 296–303.
10) Uchida, Y., Hara, M., Nishio, H., Sidransky, E., Inoue, S., Otsuka, F., Suzuki, A., Elias, P.M., Holleran, W.M., & Hamanaka, S. (2000) J. Lipid Res., 41, 2071–2082.
11) Hamanaka, S., Hara, M., Nishio, H., Otsuka, F., Suzuki, A., & Uchida, Y. (2002) J. Invest. Dermatol., 119, 416–423.
12) Holleran, W.M., Ginns, E.I., Menon, G.K., Grundmann, J.U., Fartasch, M., McKinney, C.E., Elias, P.M., & Sidransky, E. (1994) J. Clin. Invest., 93, 1756–1764.
13) Schmuth, M., Man, M.Q., Weber, F., Gao, W., Feingold, K.R., Fritsch, P., Elias, P.M., & Holleran, W.M. (2000) J. Invest. Dermatol., 115, 459–466.
14) Elias, P.M., Gruber, R., Crumrine, D., Menon, G., Williams, M.L., Wakefield, J.S., Holleran, W.M., & Uchida, Y. (2014) Biochim. Biophys. Acta, 1841, 314–318.
15) Uchida, Y. & Holleran, W.M. (2008) J. Dermatol. Sci., 51, 77–87.
16) Vasireddy, V., Wong, P., & Ayyagari, R. (2010) Prog. Retin. Eye Res., 29, 191–207.
17) Sassa, T., Ohno, Y., Suzuki, S., Nomura, T., Nishioka, C., Kashiwagi, T., Hirayama, T., Akiyama, M., Taguchi, R., Shimizu, H., Itohara, S., & Kihara, A. (2013) Mol. Cell. Biol., 33, 2787–2796.
18) Ohno, Y., Suto, S., Yamanaka, M., Mizutani, Y., Mitsutake, S., Igarashi, Y., Sassa, T., & Kihara, A. (2010) Proc. Natl. Acad. Sci. USA, 107, 18439–18444.
19) Uchida, Y., Cho, Y., Moradian, S., Kim, J., Nakajima, K., Crumrine, D., Park, K., Ujihara, M., Akiyama, M., Shimizu, H., Holleran, W.M., Sano, S., & Elias, P.M. (2010) J. Invest. Dermatol., 130, 2497–2499.
20) Goto-Inoue, N., Hayasaka, T., Zaima, N., Nakajima, K., Holleran, W.M., Sano, S., Uchida, Y., & Setou, M. (2012) PLoS ONE, 7, e49519.
21) Fischer, J., Lefèvre, C., Morava, E., Mussini, J.M., Laforêt, P., Negre-Salvayre, A., Lathrop, M., & Salvayre, R. (2007) Nat. Genet., 39, 28–30.
22) Chiba, T., Thomas, C.P., Calcutt, M.W., Boeglin, W.E., O’Donnell, V.B., & Brash, A.R. (2016) J. Biol. Chem., 291, 14540–14554.
23) Alderson, N.L., Rembiesa, B.M., Walla, M.D., Bielawska, A., Bielawski, J., & Hama, H. (2004) J. Biol. Chem., 279, 48562–48568.
24) Uchida, Y., Hama, H., Alderson, N.L., Douangpanya, S., Wang, Y., Crumrine, D.A., Elias, P.M., & Holleran, W.M. (2007) J. Biol. Chem., 282, 13211–13219.
25) Kruer, M.C., Paisán-Ruiz, C., Boddaert, N., Yoon, M.Y., Hama, H., Gregory, A., Malandrini, A., Woltjer, R.L., Munnich, A., Gobin, S., Polster, B.J., Palmeri, S., Edvardson, S., Hardy, J., Houlden, H., & Hayflick, S.J. (2010) Ann. Neurol., 68, 611–618.
26) Kota, V. & Hama, H. (2014) Adv. Biol. Regul., 54, 223–230.
27) Calleja, C., Messaddeq, N., Chapellier, B., Yang, H., Krezel, W., Li, M., Metzger, D., Mascrez, B., Ohta, K., Kagechika, H., Endo, Y., Mark, M., Ghyselinck, N.B., & Chambon, P. (2006) Genes Dev., 20, 1525–1538.
28) Rivier, M., Castiel, I., Safonova, I., Ailhaud, G., & Michel, S. (2000) J. Invest. Dermatol., 114, 681–687.
29) Man, M.Q., Choi, E.H., Schmuth, M., Crumrine, D., Uchida, Y., Elias, P.M., Holleran, W.M., & Feingold, K.R. (2006) J. Invest. Dermatol., 126, 386–392.
30) Oda, Y., Uchida, Y., Moradian, S., Crumrine, D., Elias, P.M., & Bikle, D.D. (2009) J. Invest. Dermatol., 129, 1367–1378.
31) Elias, P.M., Hatano, Y., & Williams, M.L. (2008) J. Allergy Clin. Immunol., 121, 1337–1343.
32) Elias, P.M. & Wakefield, J.S. (2011) Clin. Rev. Allergy Immunol., 41, 282–295.
33) Seguchi, T., Cui, C.Y., Kusuda, S., Takahashi, M., Aisu, K., & Tezuka, T. (1996) Arch. Dermatol. Res., 288, 442–446.
34) Palmer, C.N., Irvine, A.D., Terron-Kwiatkowski, A., Zhao, Y., Liao, H., Lee, S.P., Goudie, D.R., Sandilands, A., Campbell, L.E., Smith, F.J., O’Regan, G.M., Watson, R.M., Cecil, J.E., Bale, S.J., Compton, J.G., DiGiovanna, J.J., Fleckman, P., Lewis-Jones, S., Arseculeratne, G., Sergeant, A., Munro, C.S., El Houate, B., McElreavey, K., Halkjaer, L.B., Bisgaard, H., Mukhopadhyay, S., & McLean, W.H. (2006) Nat. Genet., 38, 441–446.
35) Elias, P.M. (2015) Exp. Dermatol., 24, 179–180.
36) Imokawa, G., Abe, A., Jin, K., Higaki, Y., Kawashima, M., & Hidano, A. (1991) J. Invest. Dermatol., 96, 523–526.
37) Bleck, O., Abeck, D., Ring, J., Hoppe, U., Vietzke, J.P., Wolber, R., Brandt, O., & Schreiner, V. (1999) J. Invest. Dermatol., 113, 894–900.
38) Janssens, M., van Smeden, J., Gooris, G.S., Bras, W., Portale, G., Caspers, P.J., Vreeken, R.J., Hankemeier, T., Kezic, S., Wolterbeek, R., Lavrijsen, A.P., & Bouwstra, J.A. (2012) J. Lipid Res., 53, 2755–2766.
39) Thakoersing, V.S., Gooris, G.S., Mulder, A., Rietveld, M., El Ghalbzouri, A., & Bouwstra, J.A. (2012) Tissue Eng. Part C Methods, 18, 1–11.
40) van Smeden, J., Janssens, M., Kaye, E.C., Caspers, P.J., Lavrijsen, A.P., Vreeken, R.J., & Bouwstra, J.A. (2014) Exp. Dermatol., 23, 45–52.
41) Higuchi, K., Hara, J., Okamoto, R., Kawashima, M., & Imokawa, G. (2000) Biochem. J., 350, 747–756.
42) Wakita, H., Matsushita, K., Nishimura, K., Tokura, Y., Furukawa, F., & Takigawa, M. (1998) J. Invest. Dermatol., 110, 253–258.
43) Sun, L., Xu, L., Henry, F.A., Spiegel, S., & Nielsen, T.B. (1996) J. Invest. Dermatol., 106, 232–237.
44) Yamakawa, T. & Iida, T. (1953) Jpn. J. Exp. Med., 23, 327–331.
45) Higashi, H. (2007) Yakugaku Zasshi, 127, 563–570.
46) Ledeen, R.W. (1984) J. Neurosci. Res., 12, 147–159.
47) Bremer, E.G., Schlessinger, J., & Hakomori, S. (1986) J. Biol. Chem., 261, 2434–2440.
48) Paller, A.S., Arnsmeier, S.L., Alvarez-Franco, M., & Bremer, E.G. (1993) J. Invest. Dermatol., 100, 841–845.
49) Paller, A.S., Arnsmeier, S.L., Fisher, G.J., & Yu, Q.C. (1995) Exp. Cell Res., 217, 118–124.
50) Wang, X.Q., Sun, P., & Paller, A.S. (2001) J. Biol. Chem., 276, 44504–44511.
51) Liu, J.W., Sun, P., Yan, Q., Paller, A.S., Gerami, P., Ho, N., Vashi, N., Le Poole, I.C., & Wang, X.Q. (2009) Cancer Res., 69, 8662–8669.
52) Randeria, P.S., Seeger, M.A., Wang, X.Q., Wilson, H., Shipp, D., Mirkin, C.A., & Paller, A.S. (2015) Proc. Natl. Acad. Sci. USA, 112, 5573–5578.
53) Uchida, Y., Iwamori, M., & Nagai, Y. (1990) Biochem. Biophys. Res. Commun., 170, 162–168.
54) Uchida, Y., Ogawa, T., Iwamori, M., & Nagai, Y. (1991) J. Biochem., 109, 462–465.
55) Clayton, R.B., Cooper, J.M., Curstedt, T., Sjövall, J., Borsook, H., Chin, J., & Schwarz, A. (1974) J. Lipid Res., 15, 557–562.
56) Okazaki, T., Bielawska, A., Bell, R.M., & Hannun, Y.A. (1990) J. Biol. Chem., 265, 15823–15831.
57) Okazaki, T., Bell, R.M., & Hannun, Y.A. (1989) J. Biol. Chem., 264, 19076–19080.
58) Obeid, L.M., Okazaki, T., Karolak, L.A., & Hannun, Y.A. (1990) J. Biol. Chem., 265, 2370–2374.
59) Kolesnick, R.N. (1989) J. Biol. Chem., 264, 7617–7623.
60) Sultan, I., Senkal, C.E., Ponnusamy, S., Bielawski, J., Szulc, Z., Bielawska, A., Hannun, Y.A., & Ogretmen, B. (2006) Biochem. J., 393, 513–521.
61) Wakita, H., Tokura, Y., Yagi, H., Nishimura, K., Furukawa, F., & Takigawa, M. (1994) Arch. Dermatol. Res., 286, 350–354.
62) Uchida, Y., Nardo, A.D., Collins, V., Elias, P.M., & Holleran, W.M. (2003) J. Invest. Dermatol., 120, 662–669.
63) Uchida, Y., Houben, E., Park, K., Douangpanya, S., Lee, Y.M., Wu, B.X., Hannun, Y.A., Radin, N.S., Elias, P.M., & Holleran, W.M. (2010) J. Invest. Dermatol., 130, 2472–2480.
64) Truman, J.P., Garcia-Barros, M., Obeid, L.M., & Hannun, Y.A. (2013) Biochim. Biophys. Acta, 1841, 1174–1188.
65) Elojeimy, S., Liu, X., McKillop, J.C., El-Zawahry, A.M., Holman, D.H., Cheng, J.Y., Meacham, W.D., Mahdy, A.E., Saad, A.F., Turner, L.S., Cheng, J., A Day, T., Dong, J.Y., Bielawska, A., Hannun, Y.A., & Norris, J.S. (2007) Mol. Ther., 15, 1259–1263.
66) Charruyer, A., Bell, S.M., Kawano, M., Douangpanya, S., Yen, T.Y., Macher, B.A., Kumagai, K., Hanada, K., Holleran, W.M., & Uchida, Y. (2008) J. Biol. Chem., 283, 16682–16692.
67) Uchida, Y. (2014) Biochim. Biophys. Acta, 1841, 453–462.
68) Kim, S., Hong, I., Hwang, J.S., Choi, J.K., Rho, H.S., Kim, D.H., Chang, I., Lee, S.H., Lee, M.O., & Hwang, J.S. (2006) Mol. Med., 12, 17–24.
69) Stoffel, W., Assmann, G., & Binczek, E. (1970) Hoppe Seylers Z. Physiol. Chem., 351, 635–642.
70) Vogler, R., Sauer, B., Kim, D.S., Schafer-Korting, M., & Kleuser, B. (2003) J. Invest. Dermatol., 120, 693–700.
71) Sauer, B., Vogler, R., von Wenckstern, H., Fujii, M., Anzano, M.B., Glick, A.B., Schäfer-Korting, M., Roberts, A.B., & Kleuser, B. (2004) J. Biol. Chem., 279, 38471–38479.
72) Kim, D.S., Kim, S.Y., Kleuser, B., Schäfer-Korting, M., Kim, K.H., & Park, K.C. (2004) Cell. Signal., 16, 89–95.
73) Schuppel, M., Kurschner, U., Kleuser, U., Schafer-Korting, M., & Kleuser, B. (2008) J. Invest. Dermatol., 128, 1747–1756.
74) Manggau, M., Kim, D.S., Ruwisch, L., Vogler, R., Korting, H.C., Schäfer-Korting, M., & Kleuser, B. (2001) J. Invest. Dermatol., 117, 1241–1249.
75) Matloubian, M., Lo, C.G., Cinamon, G., Lesneski, M.J., Xu, Y., Brinkmann, V., Allende, M.L., Proia, R.L., & Cyster, J.G. (2004) Nature, 427, 355–360.
76) Reines, I., Kietzmann, M., Mischke, R., Tschernig, T., Lüth, A., Kleuser, B., & Bäumer, W. (2009) J. Invest. Dermatol., 129, 1954–1962.
77) Seo, E.Y., Park, G.T., Lee, K.M., Kim, J.A., Lee, J.H., & Yang, J.M. (2006) J. Invest. Dermatol., 126, 1187–1189.
78) Baumer, W., Rossbach, K., Mischke, R., Reines, I., Langbein-Detsch, I., Lüth, A., & Kleuser, B. (2011) J. Invest. Dermatol., 131, 266–268.
79) Wood, S.H., Clements, D.N., Ollier, W.E., Nuttall, T., McEwan, N.A., & Carter, S.D. (2009) J. Dermatol. Sci., 55, 27–33.
80) Bajjalieh, S.M., Martin, T.F., & Floor, E. (1989) J. Biol. Chem., 264, 14354–14360.
81) Kolesnick, R.N. & Hemer, M.R. (1990) J. Biol. Chem., 265, 18803–18808.
82) Granado, M.H., Gangoiti, P., Ouro, A., Arana, L., González, M., Trueba, M., & Gómez-Muñoz, A. (2009) Cell. Signal., 21, 405–412.
83) Stahelin, R.V., Subramanian, P., Vora, M., Cho, W., & Chalfant, C.E. (2007) J. Biol. Chem., 282, 20467–20474.
84) Goldsmith, M., Daka, A., Lamour, N.F., Mashiach, R., Glucksam, Y., Meijler, M.M., Chalfant, C.E., & Zor, T. (2011) Immunol. Lett., 135, 136–143.
85) Pettus, B.J., Chalfant, C.E., & Hannun, Y.A. (2002) Biochim. Biophys. Acta, 1585, 114–125.
86) Gomez-Munoz, A., Presa, N., Gomez-Larrauri, A., Rivera, I.G., Trueba, M., & Ordoñez, M. (2016) Prog. Lipid Res., 61, 51–62.
87) Hall, K., Lee, T.H., Mechler, A.I., Swann, M.J., & Aguilar, M.I. (2014) Sci. Rep., 4, 5479.
88) Brogden, K.A. (2005) Nat. Rev. Microbiol., 3, 238–250.
89) Whitelock, J.M., Murdoch, A.D., Iozzo, R.V., & Underwood, P.A. (1996) J. Biol. Chem., 271, 10079–10086.
90) Nikaido, H. & Pages, J.M. (2012) FEMS Microbiol. Rev., 36, 340–363.
91) Nakatsuji, T. & Gallo, R.L. (2012) J. Invest. Dermatol., 132, 887–895.
92) Vandamme, D., Landuyt, B., Luyten, W., & Schoofs, L. (2012) Cell. Immunol., 280, 22–35.
93) Schauber, J., Oda, Y., Büchau, A.S., Yun, Q.C., Steinmeyer, A., Zügel, U., Bikle, D.D., & Gallo, R.L. (2008) J. Invest. Dermatol., 128, 816–824.
94) Liu, P.T., Stenger, S., Li, H., Wenzel, L., Tan, B.H., Krutzik, S.R., Ochoa, M.T., Schauber, J., Wu, K., Meinken, C., Kamen, D.L., Wagner, M., Bals, R., Steinmeyer, A., Zügel, U., Gallo, R.L., Eisenberg, D., Hewison, M., Hollis, B.W., Adams, J.S., Bloom, B.R., & Modlin, R.L. (2006) Science, 311, 1770–1773.
95) Gombart, A.F. (2009) Future Microbiol., 4, 1151–1165.
96) Wang, J.W., Hogan, P.G., Hunstad, D.A., & Fritz, S.A. (2015) Pediatr. Infect. Dis. J., 34, 544–545.
97) Park, K., Elias, P.M., Oda, Y., Mackenzie, D., Mauro, T., Holleran, W.M., & Uchida, Y. (2011) J. Biol. Chem., 286, 34121–34130.
98) Senkal, C.E., Ponnusamy, S., Bielawski, J., Hannun, Y.A., & Ogretmen, B. (2009) FASEB J.
99) Park, K., Elias, P.M., Shin, K.O., Lee, Y.M., Hupe, M., Borkowski, A.W., Gallo, R.L., Saba, J., Holleran, W.M., & Uchida, Y. (2013) Mol. Cell. Biol., 33, 752–762.
100) Park, K., Ikushiro, H., Seo, H.S., Shin, K.O., Kim, Y.I., Kim, J.Y., Lee, Y.M., Yano, T., Holleran, W.M., Elias, P., & Uchida, Y. (2016) Proc. Natl. Acad. Sci. USA, 113, E1334–E1342.
101) Alvarez, S.E., Harikumar, K.B., Hait, N.C., Allegood, J., Strub, G.M., Kim, E.Y., Maceyka, M., Jiang, H., Luo, C., Kordula, T., Milstien, S., & Spiegel, S. (2010) Nature, 465, 1084–1088.
102) Hait, N.C., Allegood, J., Maceyka, M., Strub, G.M., Harikumar, K.B., Singh, S.K., Luo, C., Marmorstein, R., Kordula, T., Milstien, S., & Spiegel, S. (2009) Science, 325, 1254–1257.
103) Harikumar, K.B., Yester, J.W., Surace, M.J., Oyeniran, C., Price, M.M., Huang, W.C., Hait, N.C., Allegood, J.C., Yamada, A., Kong, X., Lazear, H.M., Bhardwaj, R., Takabe, K., Diamond, M.S., Luo, C., Milstien, S., Spiegel, S., & Kordula, T. (2014) Nat. Immunol., 15, 231–238.
104) Kim, Y.I., Park, K., Kim, J.Y., Seo, H.S., Shin, K.O., Lee, Y.M., Holleran, W.M., Elias, P.M., & Uchida, Y. (2014) Mol. Cell. Biol., 34, 4368–4378.
105) Bernard, J.J. & Gallo, R.L. (2010) J. Immunol., 185, 6535–6544.
106) Gray, G.M., White, R.J., Williams, R.H., & Yardley, H.J. (1982) Br. J. Dermatol., 106, 59–63.
107) Elias, P.M. & Friend, D.S. (1975) J. Cell Biol., 65, 180–191.
108) Wertz, P.W. & Downing, D.T. (1983) J. Lipid Res., 24, 759–765.
109) Wertz, P.W., Downing, D.T., Freinkel, R.K., & Traczyk, T.N. (1984) J. Invest. Dermatol., 83, 193–195.
110) Abraham, W., Wertz, P.W., & Downing, D.T. (1985) J. Lipid Res., 26, 761–766.
111) Wertz, P.W. & Downing, D.T. (1983) J. Lipid Res., 24, 1135–1139.
112) Wertz, P.W. & Downing, D.T. (1983) J. Lipid Res., 24, 753–758.
113) Bowser, P.A. & White, R.J. (1985) Br. J. Dermatol., 112, 1–14.
114) Holleran, W.M., Takagi, Y., Imokawa, G., Jackson, S., Lee, J.M., & Elias, P.M. (1992) J. Lipid Res., 33, 1201–1209.
115) Matsuo, N., Nomura, T., & Imokawa, G. (1992) Biochim. Biophys. Acta, 1116, 97–103.
116) Akimoto, K., Yoshikawa, N., Higaki, Y., Kawashima, M., & Imokawa, G. (1993) J. Dermatol., 20, 1–6.
117) Imokawa, G., Yada, Y., Higuchi, K., Okuda, M., Ohashi, Y., & Kawamata, A. (1994) J. Clin. Invest., 94, 89–96.
118) Jin, K., Higaki, Y., Takagi, Y., Higuchi, K., Yada, Y., Kawashima, M., & Imokawa, G. (1994) Acta Derm. Venereol., 74, 337–340.
119) Yoshikawa, N., Imokawa, G., Akimoto, K., Jin, K., Higaki, Y., & Kawashima, M. (1994) Dermatology, 188, 207–214.
120) Yada, Y., Higuchi, K., & Imokawa, G. (1995) J. Biol. Chem., 270, 12677–12684.
121) Murata, Y., Ogata, J., Higaki, Y., Kawashima, M., Yada, Y., Higuchi, K., Tsuchiya, T., Kawainami, S., & Imokawa, G. (1996) J. Invest. Dermatol., 106, 1242–1249.
122) Takagi, Y., Kriehuber, E., Imokawa, G., Elias, P.M., & Holleran, W.M. (1999) J. Lipid Res., 40, 861–869.
123) Hara, J., Higuchi, K., Okamoto, R., Kawashima, M., & Imokawa, G. (2000) J. Invest. Dermatol., 115, 406–413.
124) Uchida, Y., Iwamori, M., & Nagai, Y. (1988) Jpn. J. Exp. Med., 58, 153–161.
125) Uchida, Y., Iwamori, M., & Nagai, Y. (1990) Biochem. Biophys. Res. Commun., 170, 162–168.
126) Hamanaka, S., Ujihara, M., Uchida, Y., & Mimura, K. (1995) Skin Res., 37, 619–625.
127) Uchida, Y., Hamanaka, S., Matsuda, K., Mimura, K., & Otsuka, F. (1996) J. Dermatol. Sci., 12, 64–68.
128) Tokudome, Y., Saito, Y., Sato, F., Kikuchi, M., Hinokitani, T., & Goto, K. (2009) Colloids Surf. B Biointerfaces, 73, 92–96.
129) Denda, M., Koyama, J., Hori, J., Horii, I., Takahashi, M., Hara, M., & Tagami, H. (1993) Arch. Dermatol. Res., 285, 415–417.
130) Denda, M., Denda, S., Tsutsumi, M., Goto, M., Kumamoto, J., Nakatani, M., Takei, K., Kitahata, H., Nakata, S., Sawabu, Y., Kobayashi, Y., & Nagayama, M. (2014) Exp. Dermatol., 23, 79–82.
131) Kobayashi, Y., Sanno, Y., Sakai, A., Sawabu, Y., Tsutsumi, M., Goto, M., Kitahata, H., Nakata, S., Kumamoto, J., Denda, M., & Nagayama, M. (2014) PLoS ONE, 9, e92650.
132) Kobayashi, Y., Sawabu, Y., Kitahata, H., Denda, M., & Nagayama, M. (2016) J. Theor. Biol., 397, 52–60.