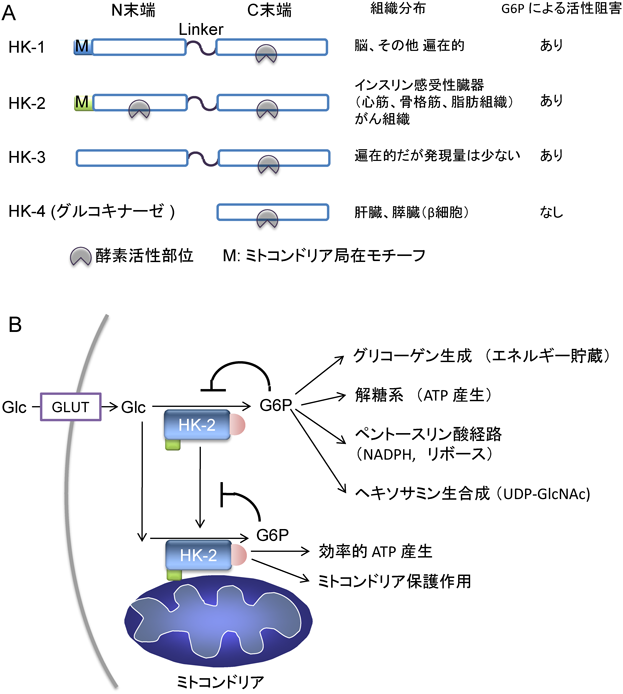
ヘキソキナーゼ(hexokinase)は解糖系の初段酵素であり,細胞内に取り込まれたグルコースをリン酸化し,グルコース6-リン酸(glucose 6-phosphate:G6P)を生成する.G6Pは解糖系のみならず,ペントースリン酸経路(pentose phosphate pathway:PPP),グリコーゲン生成,そしてヘキソサミン生合成の基質として利用される.したがって,ヘキソキナーゼは,同化作用,異化作用の両面を制御する重要な酵素といえる1–5).
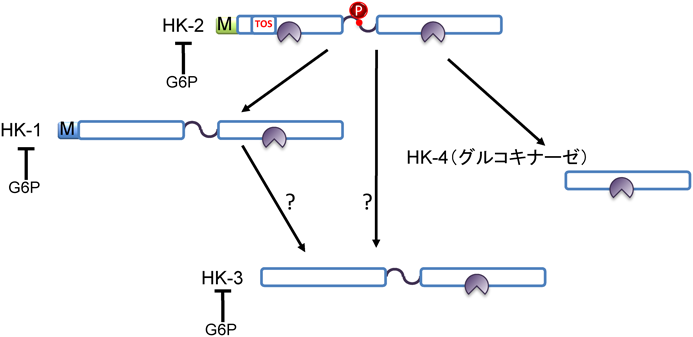
ヘキソキナーゼには四つのアイソザイムが存在する.ヘキソキナーゼ1, 2,そして3は生物の進化過程で酵母等のプロトタイプ型からの遺伝子重複と融合が起こり,相同性の高いポリペプチドが二つつながった100 kDaの分子である.一方,ヘキソキナーゼ4(グルコキナーゼ)は単量体型で50 kDaである(図1A).ヘキソキナーゼ2のみが二つの酵素活性領域の活性を保存しており,ヘキソキナーゼ1と3ではC末端側の半分が酵素活性を持つ.ヘキソキナーゼ1は脳における主要なヘキソキナーゼであるが,脳以外にもさまざまな組織に広く発現しており,ヘキソキナーゼ3はさまざまな組織に発現がみられるものの,どの組織においてもその発現は低く,ヘキソキナーゼ4は主に肝臓と膵臓に発現している.ヘキソキナーゼ2は心筋細胞,骨格筋細胞,脂肪細胞等のインスリン感受性臓器に多く発現している2–8).
1960~70年代の研究により,ヘキソキナーゼ1と2が細胞質だけではなくミトコンドリアにも局在することが見いだされ9, 10),その後の研究によりN末端がミトコンドリア局在モチーフとして重要であることが示された11, 12).このミトコンドリアへの局在は,解糖系と酸化的リン酸化の共役(カップリング)を促進するというエネルギー代謝における利点とミトコンドリア保護作用という二つの利点を併せ持つ(図1B)2, 4, 13–15).ヘキソキナーゼ1, 2と3の活性は,G6Pによる負のフィードバック制御を受けることが知られているが,ヘキソキナーゼ1と2のミトコンドリア局在は,その活性同様にG6Pによる負の制御を受けることがin vitro実験において示されている9, 16–20).しかし,細胞内ではヘキソキナーゼ2の局在が虚血状態や増殖因子の存在等の細胞内外の環境に応じて大きく変化するのに対して,ヘキソキナーゼ1の局在は比較的一定で変化しにくく,実際のヘキソキナーゼ1とヘキソキナーゼ2のミトコンドリア局在には生理学的な違いがある17, 21–23).
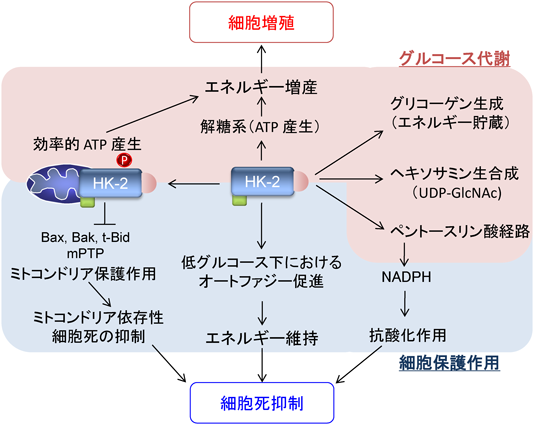
ヘキソキナーゼ2は多くのがん細胞で過剰発現がみられ,がん細胞の特徴の一つであるWarburg効果24),すなわち有酸素下においてがん細胞がミトコンドリアの酸化的リン酸化よりも,解糖系を用いてアデノシン5′-三リン酸(adenosine 5′-triphosphate:ATP)を産生する現象[好気的解糖(aerobic glycolysis)]への関与が指摘されている15, 25–28).がん細胞の増殖に伴い,他のアイソザイムからヘキソキナーゼ2へのシフトも報告されている.たとえば,正常な肝細胞における主なヘキソキナーゼアイソザイムはヘキソキナーゼ4(グルコキナーゼ)であり,ヘキソキナーゼ2の発現はみられない.しかし,肝細胞がんにおいては発現パターンの逆転が起こり,ヘキソキナーゼ4の発現が抑えられ,ヘキソキナーゼ2の発現増加がみられる29, 30).同様にヘキソキナーゼ1からヘキソキナーゼ2へのシフトがグリア細胞から多形性膠芽腫への腫瘍形成で報告されている31).がん細胞における代謝系の再プログラム化(metabolic reprogramming)の重要性が注目を集めているが,ヘキソキナーゼ2の過剰発現もその一因として捉えることができる.エネルギー代謝における重要性のみならず,ヘキソキナーゼ2はその多様な細胞保護作用(図2)を介して,がん細胞死抑制にも重要な役割を担っている.ヘキソキナーゼ2コンディショナルノックアウトマウスを使った最近の研究において,ヘキソキナーゼ2ががんの発生の初期段階(initiation)からその維持(maintenance)に至る過程において重要な役割を果たしていることが示され,がん治療のターゲット分子として注目されている32).
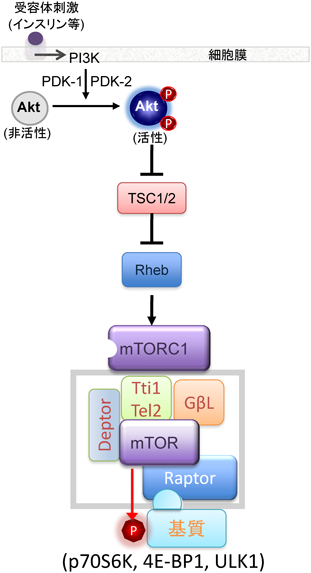
Aktは,インスリンによりホスホイノシチド3-キナーゼ(phosphoinositide 3-kinase:PI3K)を介して活性化される酵素であり,細胞膜上の糖輸送体(glucose transporter:GLUT)を増加させることでインスリンによるグルコースの細胞内への取り込みの増加に重要な役割を果たしている(図3)3, 33–39).
Aktはインスリンのみならず,さまざまなチロシンキナーゼ型受容体やGタンパク質共役受容体(GPCR)刺激に応答して活性化されることが知られている.グルコースの取り込みの増加作用に加えて,Aktは細胞保護作用や細胞増殖作用を有しており,がん細胞での過剰発現と活性化が知られている3, 33, 34).たとえば,Aktの活性化は,EGF受容体の活性化変異や,PTEN遺伝子変異によるがん発症の一因である.Aktの下流の中心的シグナル分子として哺乳類ラパマイシン標的タンパク質(mammalian target of rapamycin:mTOR)がある(図3).mTORは,機能的に異なる2種類のタンパク質複合体(mTORC1とmTORC2)を形成し,regulatory associated protein of mTOR(Raptor)を含む複合体がmTORC1, rapamycin-insensitive companion of mTOR(Rictor)を含む複合体がmTORC2である40–42).mTORC1は,p70S6キナーゼ(p70S6K)や4E-BP1などの制御を介して細胞増殖を促進し,ULK1を阻害することでオートファジーに対するブレーキとして機能している.
エネルギー代謝シグナルと細胞生存シグナルのクロストークは,細胞内外における環境変化への迅速な細胞応答を可能にする.本稿では,その一例としてヘキソキナーゼ2とAkt/mTORC1シグナル伝達経路の相互作用によるエネルギー代謝の調節,ミトコンドリア保護作用,そしてオートファジーの制御について筆者らの研究成果を交えつつ,最近の知見を紹介する(図2).
これまでに,さまざまな細胞においてヘキソキナーゼ2の強制発現による細胞保護作用が報告されている(図2).たとえば,我々は心筋細胞においてヘキソキナーゼ2のアデノウイルスによる発現が活性酸素種(reactive oxygen species:ROS)による細胞死を抑制することを示したし20, 76),多形性膠芽腫においてもヘキソキナーゼ2の強制発現がその増殖能の増強と細胞死の抑制を引き起こすことが示されている31).心特異的ヘキソキナーゼ2トランスジェニックマウスでは圧負荷誘発性の心肥大が抑制され77),一方,ヘキソキナーゼ2のヘテロ欠損マウスでは肥大の進行と心不全への移行が促進される78).活性酸素の増加はさまざまな病態の誘発因子であるが,ヘキソキナーゼ2の心保護作用の一つの機序として,ペントースリン酸経路を介してのNADPHの増加による抗酸化作用があげられる(図2)77, 78).GLUT1とヘキソキナーゼ1の発現による造血細胞における細胞保護作用や,TIGAR(TP53-induced glycolysis and apoptosis regulator)による細胞保護作用にもペントースリン酸経路の増強によるNADPHの増加が重要な役割を果たすことが知られている79, 80).
ミトコンドリアはATPを合成するエネルギー生産工場という生理学的な役割を担う一方,ストレス条件下ではアポトーシスやネクローシスを誘発することで細胞死をつかさどる(mitochondrial death pathway).ミトコンドリアに局在するヘキソキナーゼ2は,このmitochondrial death pathwayに対して抑制的に働く(図2).ミトコンドリアによるアポトーシスの促進は,アポトーシス促進性のBcl-2ファミリー(Bax, Bak, t-Bid等)がミトコンドリア外膜に細孔を形成し,その外膜透過性を上げることで引き起こされるが81–84),ヘキソキナーゼ2は,Bax, Bak, t-Bidのミトコンドリア外膜への局在を拮抗的に阻害することで,その細胞保護作用を示す85–90).その後,ヘキソキナーゼ2のミトコンドリアへの局在が細胞内のCa2+過負荷やROSによる細胞死を抑制することが示された86).ミトコンドリア膜透過性遷移孔(mitochondrial permeability transition pore:mPTP)の開口は,細胞内Ca2+過負荷やROSの増加によって引き起こされ,1.5 kDa以下の分子を非選択的に通過させ,ミトコンドリア内膜の脱分極,膨張と外膜の破裂,最終的にはネクローシスやアポトーシスを惹起する82, 91–93).したがって,先の実験結果は,ヘキソキナーゼ2のミトコンドリアへの局在がmPTPの開口を阻害することを示唆するものであり,その後この抗mPTP作用は,我々を含む多数のその後の研究によって,心筋細胞やさまざまながん細胞で重要な役割を果たすことが確認された76, 94–97).しかし,mPTPの正確な構成分子がいまだに同定されていないこともあり,その作用機序は完全には明らかになってはいない.前述のヘキソキナーゼ2のミトコンドリアへの局在による解糖系と酸化的リン酸化の共役の促進が,結果的にミトコンドリア内膜によるプロトン勾配を減少させることで内膜をわずかに脱分極させ,ミトコンドリアによるROSの産生を抑制することでmPTPの開口を抑えることが示唆されている98, 99).わずかな脱分極によるROS産生抑制と保護作用は,ミトコンドリアATP感受性K+(KATP)チャネル開口薬の細胞保護作用100, 101)やミトコンドリアタンパク質である脱共役タンパク質(uncoupling protein:UCP)による細胞保護作用の作用機序102)としても知られている.
しかしながら,ヘキソキナーゼ2のミトコンドリアへの局在の増加は,過酸化水素によるmPTPの開口とその結果としての細胞死を抑制することから20, 76, 103),より直接的な阻害作用の存在も示唆される.ノックアウトマウスを用いた実験より,cyclophilin DはmPTPの開口を促進する分子として作用することが示されており104, 105),ヘキソキナーゼ2(抑制性)とcyclophilin D(促進性)の両分子がどのように機能的に関連し,またどのようにそれが制御されているのか解明が待たれる.これまでadenine nucleotide translocase(ANT),voltage-dependent anion channel(VDAC)やmitochondrial phosphate carrier(PiC)といったさまざまな分子がmPTPの構成分子として示唆されてきたが,ノックアウトマウスの解析によりその中心的な分子としての関与が否定されてきた93, 106–109).現在,ATP合成酵素(F1Fo ATP synthase)がmPTPの主要な構成分子として有力視されており110),ヘキソキナーゼ2を含むmPTPの制御分子との物理的・機能的関係性の研究が進んでいる93, 111).また,アポトーシス促進性のBcl-2ファミリータンパク質(Bax/Bak)によるmPTPの制御も議論に上がって久しいが,最近の研究により,Bax/BakがmPTPの感受性を増加させることが報告された112).また,Bax/Bakによるミトコンドリア外膜の透過性の増加が,mPTPによる最終的なミトコンドリアの破裂と細胞死の誘発に関わっていることも報告された113).したがって,前述のミトコンドリアに局在したヘキソキナーゼ2によるBax/Bakに対する競合的な拮抗作用が,mPTPによる細胞死への抑制につながる可能性がある.
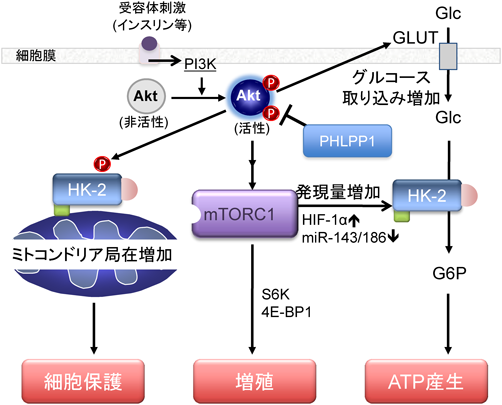
4. Aktによるヘキソキナーゼ2のミトコンドリア局在の制御
Aktは,グルコース細胞内取り込みの促進作用とは別に,強い細胞保護作用を持つリン酸化酵素である3, 33–39).Aktのミトコンドリア保護作用としては,アポトーシス促進性のBH3-onlyタンパク質Badのリン酸化によりミトコンドリア外膜透過性の上昇を抑制することがよく知られているが114),2000年代初頭に,Aktの過剰発現によってミトコンドリア分画中のヘキソキナーゼ2活性が上昇し,この増加がAktのアポトーシス抑制作用に重要であることが示され115),続いてAktによるmPTPの阻害にもミトコンドリアに局在するヘキソキナーゼ2が中心的な役割を果たしていることが示された86).実際,心筋において,ミトコンドリアに局在するヘキソキナーゼ2のレベルはインスリンやモルヒネの投与,虚血プレコンディショニング等の虚血・再灌流障害に対して心保護作用のある処置によって増加する20, 76, 116–118).このとき,ヘキソキナーゼ1のミトコンドリア局在は変化しない.このAktによるヘキソキナーゼ2の細胞質からミトコンドリアへの移動(translocation)の機序は不明であった.我々は,心筋における虚血再灌流障害に対して強い保護作用を示すAktのミトコンドリア保護作用機序の解明を進めている中で,ヘキソキナーゼ2に注目し,ヘキソキナーゼ2がAktによるリン酸化コンセンサス配列(RxRxxS/T)を有し,Aktがヘキソキナーゼ2のアミノ酸配列の473番目のトレオニン(RARQKT473)を直接リン酸化することで,ヘキソキナーゼ2のミトコンドリアへの局在を増加させ心筋細胞死を抑制することを明らかにした(図4)20, 76).その後,他の研究によっても同様な結果が示された119–123).ヘキソキナーゼ2にみられたコンセンサス配列は,ヒト,マウス,ラットと異なる種で保存されている一方,他の三つのヘキソキナーゼにはみられなかったため,ヘキソキナーゼ2に限られた制御でありAktとヘキソキナーゼ2の機能的関連性の高さを示すものと思われる.前述したように,G6Pはヘキソキナーゼ2をミトコンドリアから解離させる作用があるが,我々は,Aktによるリン酸化によりG6Pによる解離作用に対するヘキソキナーゼ2の感受性の低下が起こることを示した20).Aktによるリン酸化は,G6Pによるヘキソキナーゼ2の酵素活性阻害には影響を与えなかったことから,G6Pのヘキソキナーゼ2への結合には影響がないと思われる.ヘキソキナーゼ2は,N末端半分,C末端半分と相同性の高いポリペプチドが連結されている構造を持っており,Aktリン酸化部位であるトレオニン473は,その連結部位にあたることから,リン酸化による構造変化がミトコンドリアへの結合を強めている可能性がある.
PH domain and leucine rich repeat protein phosphatase(PHLPP)は,Aktを特異的に脱リン酸化し不活化するホスファターゼとして最近発見された124).PHLPP1とPHLPP2の二つのアイソザイムが存在し,多くのがん組織でPHLPP1またはPHLPP2の発現が減少していることから,がんにおけるAktの活性化増強の一因であると現在考えられている124–131).我々は,PHLPP1ノックアウトマウスを用いて,PHLPP1が心筋細胞に発現していること,Aktを脱リン酸化し活性を抑えることを見いだした(図4)121).また,PHLPP1は細胞質だけでなくミトコンドリアにも局在し,ミトコンドリアにおけるAktの活性そしてヘキソキナーゼ2の結合を局所的に抑制する負の制御機構として機能している可能性を示唆した121).これは,ヘキソキナーゼ2のミトコンドリアへの局在とその保護作用が,リン酸化酵素と脱リン酸化酵素のダイナミックなバランスで調節されていることを示しており,ヘキソキナーゼ2のミトコンドリア局在の調節が,さまざまな刺激に対する細胞応答の一つとして重要な役割を果たす可能性を示唆するものである.
Aktによるリン酸化以外のヘキソキナーゼ2のミトコンドリア局在調節シグナルもここで簡単にふれておきたい.1)グリコーゲン合成酵素キナーゼ(glycogen synthase kinase 3:GSK-3)は,グリコーゲン合成酵素をリン酸化し不活化する酵素として同定された.GSK-3は恒常的に活性化状態にあり,Aktによるリン酸化で不活化される132, 133).二つあるアイソザイムの一つであるGSK-3βは細胞死への関与が指摘されている分子であり,VDACをリン酸化することでヘキソキナーゼ2のVDACへの結合を阻害する.したがって,Aktの下流シグナルとして,GSK-3βがヘキソキナーゼ2のミトコンドリアへの局在を調節している可能性が示唆されている92, 111, 134).2)myotonic dystrophy protein kinase(DMPK)は,その遺伝子変異が1型筋強直性ジストロフィーの原因となる酵素であり,そのアイソザイムの一つであるDMPK-Aは,骨格筋のミトコンドリアにおいてSrcとヘキソキナーゼ2の分子複合体を形成し,ヘキソキナーゼ2のミトコンドリア局在を増加させることが報告された135).3)がん抑制遺伝子であるp53は細胞死を促進する転写因子であるが,低レベルでの活性化や生理学的なレベルの活性化では逆に抗酸化作用を示す細胞保護的な側面があることが報告されている.前述のTIGARはp53によってその発現が調節される分子であり,ペントースリン酸経路の促進による抗酸化作用79, 80)を示すが,同時にミトコンドリアにも局在し,ヘキソキナーゼ2のミトコンドリアへの結合を安定化することでミトコンドリアによるROSの産生を抑えることが示された136).
最近,マクロファージにおけるグラム陽性菌の細胞壁の一部をなす多糖であるペプチドグリカンによるNLRP3インフラマソーム(inflammasome)の活性化にヘキソキナーゼ2のミトコンドリアからの遊離が必要であること,ヘキソキナーゼ2のミトコンドリアからの遊離がNLRP3インフラマソームを活性化するのに十分であることが示された137).ヘキソキナーゼ2のミトコンドリア局在・細胞内局在の変化が,自然免疫応答(innate immune response)を調節することを示す結果である.以前から,がん細胞同様,免疫細胞の活性化においてヘキソキナーゼ2発現増加と好気的解糖促進が起こることが報告されており138–140),免疫細胞におけるmetabolic reprogrammingや,その機能へのヘキソキナーゼ2の関与は今後の楽しみな研究領域である.また,ヘキソキナーゼ2のミトコンドリア局在の減少そのものが細胞内シグナルとして機能することは,ヘキソキナーゼ2のミトコンドリアへの局在が虚血心筋で減少していること23, 141, 142)を考え合わせてみると非常に興味深い.
5. ヘキソキナーゼ2によるmTORC1の抑制とオートファジーの調節
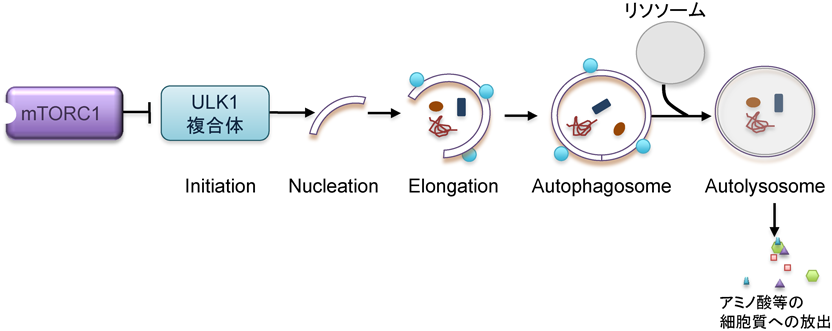
オートファジー(自食作用)は,50年ほど前にリソソームの発見者であるDe Duve博士によって最初に報告・命名された細胞機能であり,栄養飢餓状態に反応して活性化される143).オートファジーは,細胞内の不要な分子や細胞内小器官をオートファゴソーム(autophagosome)と呼ばれる小胞に取り込み,リソソームと融合・消化することでアミノ酸や脂肪酸を作り出し,栄養飢餓状態でのタンパク質の合成やエネルギー産生を可能にして細胞生存を延長する144–146).オートファジーは,細胞がエネルギーの不足を感知して起こり以下の段階を経て完了する(図5).1)細胞質中に膜構造が形成される(membrane nucleation),2)膜の伸張と細胞質成分の取り込み(elongation),3)二重膜からなる小胞の形成(autophagosome formation),4)オートファゴソームのリソソームとの融合(autolysosome formation),5)リソソームによる内容物の分解である.オートファジーの発見後,それが酵母からヒトまで真核生物に広く保存された細胞機能であることが明らかとなったが,その分子機構や生理的意義は長らく明確になっていなかった.オートファジーによる細胞内での基質分解の分子制御の解明が進んだのは発見から30年ほどの月日を経てからのことであり,大隅博士による出芽酵母におけるautophagy-related(Atg)遺伝子の発見による147, 148).現在では,30以上ものAtg遺伝子が同定されており,これらの分子によってオートファジーの一連の過程が厳密に制御されていることがわかっている149).続いて,細胞が栄養飢餓状態を感知し,オートファジーの機構へシグナルを伝達する上で重要な役割を果たすのがmTORC1であることが解明された150–154).細胞増殖因子の存在下や富栄養状態において活性化されるmTORC1は,ATG1(ULK1)をリン酸化しその活性を抑制することでオートファジーの開始を抑制している155–161).一方,虚血等の細胞増殖因子やエネルギー代謝基質の細胞への供給が制限されている状態では,mTORC1の上流分子であるAktの活性の減少,また,ATPの減少(AMP/ATP比の増加)によって活性化されるAMP活性化プロテインキナーゼ(AMPK)の活性化がmTORC1の抑制そしてULK1の活性化を引き起こし,オートファジーを惹起する158, 161–165).
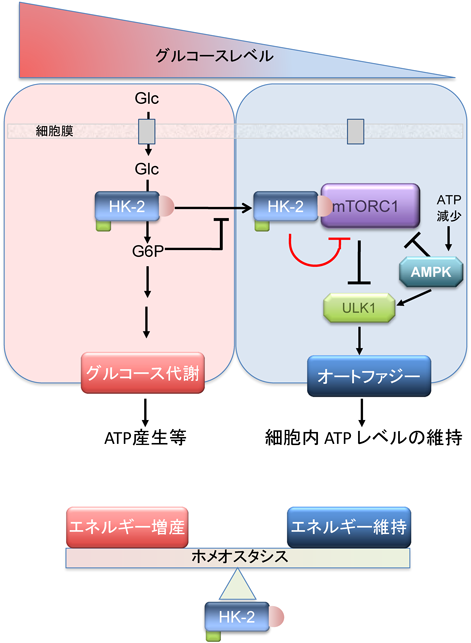
近年の研究により,アミノ酸の増加が直接mTORC1をリソソームにおいて活性化することが明らかになってきたが151, 166–174),主要な細胞エネルギー源の一つであるグルコースの代謝とオートファジーの分子的な関連は報告されていなかった.エネルギー代謝系とmTORC1を含むオートファジー分子機構との直接的な分子間相互作用の存在は,虚血等の栄養飢餓状態に対する迅速なオートファジーの開始を可能とする利点があるはずである.最近,我々はヘキソキナーゼ2がその基質であるグルコース非存在下において,mTORC1を阻害してオートファジーを促進させることを示した(図6)175).この予期せぬヘキソキナーゼ2の作用を発見した最初のきっかけは,心筋細胞においてグルコース非存在下で活性化されたオートファジーが,グルコースのアナログである2-デオキシ-D-グルコース(2-DG)の投与によって阻害されるという観察結果であった.2-DGは,細胞内に取り込まれヘキソキナーゼに結合してリン酸化を受けるものの,その後のATP産生の代謝経路には利用されないため,解糖系阻害剤として実験に使われ,またがん治療薬としての可能性も示唆されている.この“グルコース非存在下におけるオートファジーのグルコースアナログによる抑制”という結果から,解糖系によるATP産生への寄与とは別に,ヘキソキナーゼ2がオートファジーの制御に直接関連している可能性が示唆されたのである.事実,ヘキソキナーゼ2の過剰発現はグルコース非存在下でのオートファジーと細胞の生存を増加させ,ヘキソキナーゼ2ノックダウンはオートファジーと細胞の生存を抑制した175).一方,ヘキソキナーゼ1のノックダウンはオートファジーにも細胞の生存にも影響を与えなかった.以上のことは,ヘキソキナーゼ2がグルコースの有無に応じて,その役割を細胞内エネルギー生産増強から最低限のエネルギーの保存へと変換しうることを示唆している175).
このヘキソキナーゼ2の細胞保護的なオートファジー促進作用は,ヘキソキナーゼ2酵素活性とミトコンドリアへの局在には依存しなかった175).したがって,ヘキソキナーゼ2のこの作用はミトコンドリア局在による細胞保護とは独立した細胞保護作用であり(図2),グルコース非存在下において発現するscaffold(足場)機能であることが推測された.いくつかの実験を通し,ヘキソキナーゼ2がグルコース非存在下において,mTORC1の活性を抑制していることが明らかになり,我々は,ヘキソキナーゼ2がmTORC1に結合し,その活性を抑えるという仮説を立てた.事実,ヘキソキナーゼ2のmTORC1の結合はグルコース非存在下において増加し,その結合は,Raptorを介したものであることが示された.我々は,ヘキソキナーゼ2のアミノ酸配列にmTOR signaling motif(TOS motif)が存在することを見いだした(アミノ酸199~203,FDIDI).TOS motifは,S6Kや4E-BP1などmTORC1の基質にみられるモチーフ配列であり,この配列を介して基質はRaptorに認識され結合し,続いてmTORによるリン酸化を受ける176–178).そこで我々は,TOS motif変異のヘキソキナーゼ2を発現させてもmTORC1に結合せず,オートファジーの促進作用もみられないことから,ヘキソキナーゼ2はグルコース非存在下(すなわち自身の基質不在下において),mTORC1にdecoy substrate(おとり・偽の基質)として結合し,その機能を抑制していると結論づけた175).したがって,前述したように富栄養状態では,mTORC1がヘキソキナーゼ2の発現量に対して正の制御を行う一方,栄養飢餓状態では逆に,ヘキソキナーゼ2がmTORC1を抑制することで,細胞増殖を抑制,細胞保護的なオートファジーを促進させるという相互作用が示唆された(図6)175, 179).
では,ヘキソキナーゼ2のグルコース代謝とオートファジー促進の二つの機能のスイッチは何か? この問いに対する答えは,完全に明確とはなっていないが,2-DGと異なり,5-チオグルコース(ヘキソキナーゼに結合するが,リン酸化されないグルコースアナログ)は,オートファジーに抑制効果をまったく示さなかったことから,グルコースのヘキソキナーゼ2に対する結合ではなく,その後のヘキソキナーゼ2による基質のリン酸化がこのスイッチに重要であることが示唆された.このことは,kinase-dead変異のヘキソキナーゼ2がグルコース存在下においてもオートファジーを誘発するが,野生型のヘキソキナーゼ2はグルコース存在下ではそのような作用を持たない,という実験結果からも支持された.すなわち,グルコース存在下ではヘキソキナーゼ2の活性産物であるG6Pがヘキソキナーゼ2のmTORC1に対する阻害性の結合を抑制しており,この抑制がグルコース非存在下で解除されることで,mTORC1の阻害・オートファジーの促進へと切り替わるものと考えられる.言い換えるならば,ヘキソキナーゼ2は,グルコースの供給の多寡をG6Pのレベルを介して感知し,解糖系によるATP生成とオートファジーによるエネルギーの維持を調節していることになる.また,G6Pがヘキソキナーゼ2の酵素活性,ミトコンドリア局在,そしてオートファジーへの移行のいずれをも負のネガティブフィードバック機構として制御していることは,細胞がグルコースのレベルを軸に解糖系をはじめとするさまざまなグルコース代謝,ミトコンドリアでのヘキソキナーゼ2によるATP産生促進・細胞保護作用,mTORC1による増殖シグナル,オートファジーを協調的に調整していることを意味し,エネルギー代謝と増殖等の細胞機能との密接かつ直接的な関係を示唆するものであると思われる.
脂肪酸はミトコンドリアにおけるATP産生にきわめて重要な基質であるが,脂肪酸の欠乏とmTORC1抑制を介したオートファジーの関係については,不明なことが多く今後の課題といえる.ただ,最近の研究により,栄養飢餓状態で活性化されたオートファジーが,細胞の膜成分を分解することで,脂肪滴(lipid droplet)へ脂肪酸を供給し,続いて脂肪分解(lipolysis)によって得られた脂肪酸がミトコンドリアに輸送されることが示された180).これは,オートファジーとミトコンドリアにおける酸化的リン酸化が連携することで,栄養飢餓状態でのATP産生を進めていることを示している.またこのとき,fusion(融合)によるミトコンドリア・ネットワークの形成が,輸送された脂肪酸の効果的な利用とATPの産生,ひいては細胞全体におけるエネルギー状態の改善に必要であることが示唆され180),ミトコンドリアのfusion/fission(融合と分裂)のバランス制御が,オートファジーによる細胞栄養状態の改善に重要な役割を果たすことが明らかになったといえる.
引用文献References
1) Ardehali, H., Printz, R.L., Whitesell, R.R., May, J.M., & Granner, D.K. (1999) J. Biol. Chem., 274, 15986–15989.
2) Pedersen, P.L. (2007) J. Bioenerg. Biomembr., 39, 211–222.
3) Robey, R.B. & Hay, N. (2006) Oncogene, 25, 4683–4696.
4) Wilson, J.E. (1995) Rev. Physiol. Biochem. Pharmacol., 126, 65–198.
5) Wilson, J.E. (2003) J. Exp. Biol., 206, 2049–2057.
6) Katzen, H.M. & Schimke, R.T. (1965) Proc. Natl. Acad. Sci. USA, 54, 1218–1225.
7) Tsai, H.J. & Wilson, J.E. (1996) Arch. Biochem. Biophys., 329, 17–23.
8) Heikkinen, S., Suppola, S., Malkki, M., Deeb, S.S., Janne, J., & Laakso, M. (2000) Mamm. Genome, 11, 91–96.
9) Mayer, S.E., Mayfield, A.C., & Haas, J.A. (1966) Mol. Pharmacol., 2, 393–405.
10) Rose, I.A. & Warms, J.V. (1967) J. Biol. Chem., 242, 1635–1645.
11) Sui, D. & Wilson, J.E. (1997) Arch. Biochem. Biophys., 345, 111–125.
12) Xie, G.C. & Wilson, J.E. (1988) Arch. Biochem. Biophys., 267, 803–810.
13) Arora, K.K. & Pedersen, P.L. (1988) J. Biol. Chem., 263, 17422–17428.
14) Nelson, B.D. & Kabir, F. (1986) Biochimie, 68, 407–415.
15) Pedersen, P.L., Mathupala, S., Rempel, A., Geschwind, J.F., & Ko, Y.H. (2002) Biochim. Biophys. Acta, 1555, 14–20.
16) Aubert-Foucher, E., Font, B., & Gautheron, D.C. (1984) Arch. Biochem. Biophys., 232, 391–399.
17) John, S., Weiss, J.N., & Ribalet, B. (2011) PLoS One, 6, e17674.
18) Kabir, F. & Wilson, J.E. (1993) Arch. Biochem. Biophys., 300, 641–650.
19) Mathupala, S.P., Ko, Y.H., & Pedersen, P.L. (2006) Oncogene, 25, 4777–4786.
20) Roberts, D.J., Tan-Sah, V.P., Smith, J.M., & Miyamoto, S. (2013) J. Biol. Chem., 288, 23798–23806.
21) Vogt, C., Yki-Jarvinen, H., Iozzo, P., Pipek, R., Pendergrass, M., Koval, J., Ardehali, H., Printz, R., Granner, D., Defronzo, R., & Mandarino, L. (1998) J. Clin. Endocrinol. Metab., 83, 230–234.
22) Gurel, E., Ustunova, S., Kapucu, A., Yilmazer, N., Eerbeek, O., Nederlof, R., Hollmann, M.W., Demirci-Tansel, C., & Zuurbier, C.J. (2013) Mol. Biol. Rep., 40, 4153–4160.
23) Pasdois, P., Parker, J.E., Griffiths, E.J., & Halestrap, A.P. (2011) Biochem. J., 436, 493–505.
24) Warburg, O., Dickens, F., Kaiser, B., & Wilhelm-Institut fur Biologie, B. (1930) The Metabolism of Tumours: Investigations from the Kaiser-Wilhelm Institute for Biology, Constable & Co, Ltd., London.
25) Bustamante, E., Morris, H.P., & Pedersen, P.L. (1981) J. Biol. Chem., 256, 8699–8704.
26) Mathupala, S.P., Rempel, A., & Pedersen, P.L. (1997) J. Bioenerg. Biomembr., 29, 339–343.
27) Mayer, D., Klimek, F., Rempel, A., & Bannasch, P. (1997) Biochem. Soc. Trans., 25, 122–127.
28) Rempel, A., Mathupala, S.P., Griffin, C.A., Hawkins, A.L., & Pedersen, P.L. (1996) Cancer Res., 56, 2468–2471.
29) Goel, A., Mathupala, S.P., & Pedersen, P.L. (2003) J. Biol. Chem., 278, 15333–15340.
30) Kwee, S.A., Hernandez, B., Chan, O., & Wong, L. (2012) PLoS One, 7, e46591.
31) Wolf, A., Agnihotri, S., Micallef, J., Mukherjee, J., Sabha, N., Cairns, R., Hawkins, C., & Guha, A. (2011) J. Exp. Med., 208, 313–326.
32) Patra, K.C., Wang, Q., Bhaskar, P.T., Miller, L., Wang, Z., Wheaton, W., Chandel, N., Laakso, M., Muller, W.J., Allen, E.L., Jha, A.K., Smolen, G.A., Clasquin, M.F., Robey, R.B., & Hay, N. (2013) Cancer Cell, 24, 213–228.
33) Franke, T.F., Yang, S.I., Chan, T.O., Datta, K., Kazlauskas, A., Morrison, D.K., Kaplan, D.R., & Tsichlis, P.N. (1995) Cell, 81, 727–736.
34) Scheid, M.P. & Woodgett, J.R. (2001) Nat. Rev. Mol. Cell Biol., 2, 760–768.
35) Burgering, B.M. & Coffer, P.J. (1995) Nature, 376, 599–602.
36) Matsui, T., Tao, J., del Monte, F., Lee, K.H., Li, L., Picard, M., Force, T.L., Franke, T.F., Hajjar, R.J., & Rosenzweig, A. (2001) Circulation, 104, 330–335.
37) Miyamoto, S., Murphy, A.N., & Brown, J.H. (2009) J. Bioenerg. Biomembr., 41, 169–180.
38) Shiojima, I., Yefremashvili, M., Luo, Z., Kureishi, Y., Takahashi, A., Tao, J., Rosenzweig, A., Kahn, C.R., Abel, E.D., & Walsh, K. (2002) J. Biol. Chem., 277, 37670–37677.
39) Sussman, M.A., Volkers, M., Fischer, K., Bailey, B., Cottage, C.T., Din, S., Gude, N., Avitabile, D., Alvarez, R., Sundararaman, B., Quijada, P., Mason, M., Konstandin, M.H., Malhowski, A., Cheng, Z., Khan, M., & McGregor, M. (2011) Physiol. Rev., 91, 1023–1070.
40) Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., Tokunaga, C., Avruch, J., & Yonezawa, K. (2002) Cell, 110, 177–189.
41) Kim, D.H., Sarbassov, D.D., Ali, S.M., King, J.E., Latek, R.R., Erdjument-Bromage, H., Tempst, P., & Sabatini, D.M. (2002) Cell, 110, 163–175.
42) Sarbassov, D.D., Ali, S.M., Kim, D.-H., Guertin, D.A., Latek, R.R., Erdjument-Bromage, H., Tempst, P., & Sabatini, D.M. (2004) Curr. Biol., 14, 1296–1302.
43) Burcelin, R., Printz, R.L., Kande, J., Assan, R., Granner, D.K., & Girard, J. (1993) Am. J. Physiol., 265, E392–E401.
44) Katzen, H.M. (1966) Biochem. Biophys. Res. Commun., 24, 531–536.
45) Katzen, H.M., Soderman, D.D., & Wiley, C.E. (1970) J. Biol. Chem., 245, 4081–4096.
46) Printz, R.L., Koch, S., Potter, L.R., O’Doherty, R.M., Tiesinga, J.J., Moritz, S., & Granner, D.K. (1993) J. Biol. Chem., 268, 5209–5219.
47) Chehtane, M. & Khaled, A.R. (2010) Am. J. Physiol. Cell Physiol., 298, C1560–C1571.
48) Culbert, A.A. & Tavare, J.M. (2002) Biochim. Biophys. Acta, 1578, 43–50.
49) Duarte, A.I., Santos, P., Oliveira, C.R., Santos, M.S., & Rego, A.C. (2008) Biochim. Biophys. Acta, 1783, 994–1002.
50) Osawa, H., Sutherland, C., Robey, R.B., Printz, R.L., & Granner, D.K. (1996) J. Biol. Chem., 271, 16690–16694.
51) Vogt, C., Ardehali, H., Iozzo, P., Yki-Jarvinen, H., Koval, J., Maezono, K., Pendergrass, M., Printz, R., Granner, D., DeFronzo, R., & Mandarino, L. (2000) Metabolism, 49, 814–818.
52) Bhaskar, P.T., Nogueira, V., Patra, K.C., Jeon, S.M., Park, Y., Robey, R.B., & Hay, N. (2009) Mol. Cell. Biol., 29, 5136–5147.
53) Duvel, K., Yecies, J.L., Menon, S., Raman, P., Lipovsky, A.I., Souza, A.L., Triantafellow, E., Ma, Q., Gorski, R., Cleaver, S., Vander Heiden, M.G., MacKeigan, J.P., Finan, P.M., Clish, C.B., Murphy, L.O., & Manning, B.D. (2010) Mol. Cell, 39, 171–183.
54) Pusapati, R.V., Daemen, A., Wilson, C., Sandoval, W., Gao, M., Haley, B., Baudy, A.R., Hatzivassiliou, G., Evangelista, M., & Settleman, J. (2016) Cancer Cell, 29, 548–562.
55) Kruiswijk, F., Labuschagne, C.F., & Vousden, K.H. (2015) Nat. Rev. Mol. Cell Biol., 16, 393–405.
56) Vousden, K.H. & Ryan, K.M. (2009) Nat. Rev. Cancer, 9, 691–700.
57) Mathupala, S.P., Rempel, A., & Pedersen, P.L. (2001) J. Biol. Chem., 276, 43407–43412.
58) Riddle, S.R., Ahmad, A., Ahmad, S., Deeb, S.S., Malkki, M., Schneider, B.K., Allen, C.B., & White, C.W. (2000) Am. J. Physiol. Lung Cell. Mol. Physiol., 278, L407–L416.
59) Gwak, G.Y., Yoon, J.H., Kim, K.M., Lee, H.S., Chung, J.W., & Gores, G.J. (2005) J. Hepatol., 42, 358–364.
60) Kim, J.W., Gao, P., Liu, Y.C., Semenza, G.L., & Dang, C.V. (2007) Mol. Cell. Biol., 27, 7381–7393.
61) Sato-Tadano, A., Suzuki, T., Amari, M., Takagi, K., Miki, Y., Tamaki, K., Watanabe, M., Ishida, T., Sasano, H., & Ohuchi, N. (2013) Cancer Sci., 104, 1380–1388.
62) Jiang, B.H., Jiang, G., Zheng, J.Z., Lu, Z., Hunter, T., & Vogt, P.K. (2001) Cell Growth Differ., 12, 363–369.
63) Majumder, P.K., Febbo, P.G., Bikoff, R., Berger, R., Xue, Q., McMahon, L.M., Manola, J., Brugarolas, J., McDonnell, T.J., Golub, T.R., Loda, M., Lane, H.A., & Sellers, W.R. (2004) Nat. Med., 10, 594–601.
64) Zhong, H., Chiles, K., Feldser, D., Laughner, E., Hanrahan, C., Georgescu, M.M., Simons, J.W., & Semenza, G.L. (2000) Cancer Res., 60, 1541–1545.
65) Fang, R., Xiao, T., Fang, Z., Sun, Y., Li, F., Gao, Y., Feng, Y., Li, L., Wang, Y., Liu, X., Chen, H., Liu, X.Y., & Ji, H. (2012) J. Biol. Chem., 287, 23227–23235.
66) Gregersen, L.H., Jacobsen, A., Frankel, L.B., Wen, J., Krogh, A., & Lund, A.H. (2012) BMC Cancer, 12, 232.
67) Peschiaroli, A., Giacobbe, A., Formosa, A., Markert, E.K., Bongiorno-Borbone, L., Levine, A.J., Candi, E., D’Alessandro, A., Zolla, L., Finazzi Agro, A., & Melino, G. (2013) Oncogene, 32, 797–802.
68) Tong, A.W. & Nemunaitis, J. (2008) Cancer Gene Ther., 15, 341–355.
69) Yoshino, H., Enokida, H., Itesako, T., Kojima, S., Kinoshita, T., Tatarano, S., Chiyomaru, T., Nakagawa, M., & Seki, N. (2013) Cancer Sci., 104, 1567–1574.
70) Zhao, S., Liu, H., Liu, Y., Wu, J., Wang, C., Hou, X., Chen, X., Yang, G., Zhao, L., Che, H., Bi, Y., Wang, H., Peng, F., & Ai, J. (2013) Cancer Lett., 333, 253–260.
71) Matkovich, S.J., Hu, Y., & Dorn, G.W. 2nd. (2013) Circ. Res., 113, 62–71.
72) Li, C., Liu, Y., Liu, J., Chen, Y., Li, Z., Chen, X., Yang, K., Li, M., & Liu, Z. (2012) Head Neck Oncol., 4, 66.
73) Liu, L., Wang, Y., Bai, R., Yang, K., & Tian, Z. (2016) Oncogenesis, 5, e224.
74) Jiang, S., Zhang, H.W., Lu, M.H., He, X.H., Li, Y., Gu, H., Liu, M.F., & Wang, E.D. (2012) Cancer Res., 70, 3119–3127.
75) Ye, P., Liu, Y., Chen, C., Tang, F., Wu, Q., Wang, X., Liu, C.G., Liu, X., Liu, R., & Zheng, P. (2015) Mol. Cell, 57, 708–720.
76) Miyamoto, S., Murphy, A.N., & Brown, J.H. (2008) Cell Death Differ., 15, 521–529.
77) McCommis, K.S., Douglas, D.L., Krenz, M., & Baines, C.P. (2013) J. Am. Heart Assoc., 2, e000355.
78) Wu, R., Wyatt, E., Chawla, K., Tran, M., Ghanefar, M., Laakso, M., Epting, C.L., & Ardehali, H. (2012) EMBO Mol. Med., 4, 633–646.
79) Rathmell, J.C., Fox, C.J., Plas, D.R., Hammerman, P.S., Cinalli, R.M., & Thompson, C.B. (2003) Mol. Cell. Biol., 23, 7315–7328.
80) Bensaad, K., Tsuruta, A., Selak, M.A., Vidal, M.N., Nakano, K., Bartrons, R., Gottlieb, E., & Vousden, K.H. (2006) Cell, 126, 107–120.
81) Shimizu, S., Matsuoka, Y., Shinohara, Y., Yoneda, Y., & Tsujimoto, Y. (2001) J. Cell Biol., 152, 237–250.
82) Tait, S.W. & Green, D.R. (2013) Cold Spring Harb. Perspect. Biol., 5.
83) Wei, M.C., Zong, W.X., Cheng, E.H., Lindsten, T., Panoutsakopoulou, V., Ross, A.J., Roth, K.A., MacGregor, G.R., Thompson, C.B., & Korsmeyer, S.J. (2001) Science, 292, 727–730.
84) Yang, E., Zha, J., Jockel, J., Boise, L.H., Thompson, C.B., & Korsmeyer, S.J. (1995) Cell, 80, 285–291.
85) Gall, J.M., Wong, V., Pimental, D.R., Havasi, A., Wang, Z., Pastorino, J.G., Bonegio, R.G., Schwartz, J.H., & Borkan, S.C. (2011) Kidney Int., 79, 1207–1216.
86) Majewski, N., Nogueira, V., Bhaskar, P., Coy, P.E., Skeen, J.E., Gottlob, K., Chandel, N.S., Thompson, C.B., Robey, R.B., & Hay, N. (2004) Mol. Cell, 16, 819–830.
87) Majewski, N., Nogueira, V., Robey, R.B., & Hay, N. (2004) Mol. Cell. Biol., 24, 730–740.
88) Pastorino, J.G. & Hoek, J.B. (2003) Curr. Med. Chem., 10, 1535–1551.
89) Pastorino, J.G., Shulga, N., & Hoek, J.B. (2002) J. Biol. Chem., 277, 7610–7618.
90) Vyssokikh, M.Y., Zorova, L., Zorov, D., Heimlich, G., Jurgensmeier, J.J., & Brdiczka, D. (2002) Mol. Biol. Rep., 29, 93–96.
91) Baines, C.P. (2010) Annu. Rev. Physiol., 72, 61–80.
92) Miura, T. & Tanno, M. (2012) Cardiovasc. Res., 94, 181–189.
93) Halestrap, A.P. & Richardson, A.P. (2015) J. Mol. Cell. Cardiol., 78, 129–141.
94) Sun, L., Shukair, S., Naik, T.J., Moazed, F., & Ardehali, H. (2008) Mol. Cell. Biol., 28, 1007–1017.
95) Wu, R., Smeele, K.M., Wyatt, E., Ichikawa, Y., Eerbeek, O., Sun, L., Chawla, K., Hollmann, M.W., Nagpal, V., Heikkinen, S., Laakso, M., Jujo, K., Wasserstrom, J.A., Zuurbier, C.J., & Ardehali, H. (2011) Circ. Res., 108, 60–69.
96) Chiara, F., Castellaro, D., Marin, O., Petronilli, V., Brusilow, W.S., Juhaszova, M., Sollott, S.J., Forte, M., Bernardi, P., & Rasola, A. (2008) PLoS One, 3, e1852.
97) Smeele, K.M., Southworth, R., Wu, R., Xie, C., Nederlof, R., Warley, A., Nelson, J.K., van Horssen, P., van den Wijngaard, J.P., Heikkinen, S., Laakso, M., Koeman, A., Siebes, M., Eerbeek, O., Akar, F.G., Ardehali, H., Hollmann, M.W., & Zuurbier, C.J. (2011) Circ. Res., 108, 1165–1169.
98) da-Silva, W.S., Gomez-Puyou, A., de Gomez-Puyou, M.T., Moreno-Sanchez, R., De Felice, F.G., de Meis, L., Oliveira, M.F., & Galina, A. (2004) J. Biol. Chem., 279, 39846–39855.
99) Mailloux, R.J., Dumouchel, T., Aguer, C., deKemp, R., Beanlands, R., & Harper, M.E. (2011) Biochem. J., 437, 301–311.
100) Akao, M., O’Rourke, B., Kusuoka, H., Teshima, Y., Jones, S.P., & Marban, E. (2003) Circ. Res., 92, 195–202.
101) Murata, M., Akao, M., O’Rourke, B., & Marban, E. (2001) Circ. Res., 89, 891–898.
102) Mailloux, R.J. & Harper, M.E. (2011) Free Radic. Biol. Med., 51, 1106–1115.
103) Ahmad, A., Ahmad, S., Schneider, B.K., Allen, C.B., Chang, L.Y., & White, C.W. (2002) Am. J. Physiol. Lung Cell. Mol. Physiol., 283, L573–L584.
104) Baines, C.P., Kaiser, R.A., Purcell, N.H., Blair, N.S., Osinska, H., Hambleton, M.A., Brunskill, E.W., Sayen, M.R., Gottlieb, R.A., Dorn, G.W., Robbins, J., & Molkentin, J.D. (2005) Nature, 434, 658–662.
105) Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T., & Tsujimoto, Y. (2005) Nature, 434, 652–658.
106) Baines, C.P., Kaiser, R.A., Sheiko, T., Craigen, W.J., & Molkentin, J.D. (2007) Nat. Cell Biol., 9, 550–555.
107) Kokoszka, J.E., Waymire, K.G., Levy, S.E., Sligh, J.E., Cai, J., Jones, D.P., MacGregor, G.R., & Wallace, D.C. (2004) Nature, 427, 461–465.
108) Gutierrez-Aguilar, M., Douglas, D.L., Gibson, A.K., Domeier, T.L., Molkentin, J.D., & Baines, C.P. (2014) J. Mol. Cell. Cardiol., 72, 316–325.
109) Varanyuwatana, P. & Halestrap, A.P. (2012) Mitochondrion, 12, 120–125.
110) Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., Glick, G.D., Petronilli, V., Zoratti, M., Szabo, I., Lippe, G., & Bernardi, P. (2013) Proc. Natl. Acad. Sci. USA, 110, 5887–5892.
111) Bernardi, P., Rasola, A., Forte, M., & Lippe, G. (2015) Physiol. Rev., 95, 1111–1155.
112) Whelan, R.S., Konstantinidis, K., Wei, A.C., Chen, Y., Reyna, D.E., Jha, S., Yang, Y., Calvert, J.W., Lindsten, T., Thompson, C.B., Crow, M.T., Gavathiotis, E., Dorn, G.W. 2nd, O’Rourke, B., & Kitsis, R.N. (2012) Proc. Natl. Acad. Sci. USA, 109, 6566–6571.
113) Karch, J., Kwong, J.Q., Burr, A.R., Sargent, M.A., Elrod, J.W., Peixoto, P.M., Martinez-Caballero, S., Osinska, H., Cheng, E.H., Robbins, J., Kinnally, K.W., & Molkentin, J.D. (2013) eLife, 2, e00772.
114) Datta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., & Greenberg, M.E. (1997) Cell, 91, 231–241.
115) Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R.B., & Hay, N. (2001) Genes Dev., 15, 1406–1418.
116) Russell, R.R. 3rd, Mrus, J.M., Mommessin, J.I., & Taegtmeyer, H. (1992) J. Clin. Invest., 90, 1972–1977.
117) Southworth, R., Davey, K.A., Warley, A., & Garlick, P.B. (2007) Am. J. Physiol. Heart Circ. Physiol., 292, H378–H386.
118) Zuurbier, C.J., Eerbeek, O., & Meijer, A.J. (2005) Am. J. Physiol. Heart Circ. Physiol., 289, H496–H499.
119) Ahn, K.J., Hwang, H.S., Park, J.H., Bang, S.H., Kang, W.J., Yun, M., & Lee, J.D. (2009) J. Nucl. Med., 50, 1525–1532.
120) Betz, C., Stracka, D., Prescianotto-Baschong, C., Frieden, M., Demaurex, N., & Hall, M.N. (2013) Proc. Natl. Acad. Sci. USA, 110, 12526–12534.
121) Miyamoto, S., Purcell, N.H., Smith, J.M., Gao, T., Whittaker, R., Huang, K., Castillo, R., Glembotski, C.C., Sussman, M.A., Newton, A.C., & Brown, J.H. (2010) Circ. Res., 107, 476–484.
122) Picone, P., Giacomazza, D., Vetri, V., Carrotta, R., Militello, V., San Biagio, P.L., & Di Carlo, M. (2011) Aging Cell, 10, 832–843.
123) Rajala, A., Gupta, V.K., Anderson, R.E., & Rajala, R.V. (2013) Mitochondrion, 13, 566–576.
124) Gao, T., Furnari, F., & Newton, A.C. (2005) Mol. Cell, 18, 13–24.
125) Brognard, J., Niederst, M., Reyes, G., Warfel, N., & Newton, A.C. (2009) J. Biol. Chem., 284, 15215–15223.
126) Brognard, J., Sierecki, E., Gao, T., & Newton, A.C. (2007) Mol. Cell, 25, 917–931.
127) Gao, T., Brognard, J., & Newton, A.C. (2008) J. Biol. Chem., 283, 6300–6311.
128) Hirano, I., Nakamura, S., Yokota, D., Ono, T., Shigeno, K., Fujisawa, S., Shinjo, K., & Ohnishi, K. (2009) J. Biol. Chem., 284, 22155–22165.
129) Liu, J., Weiss, H.L., Rychahou, P., Jackson, L.N., Evers, B.M., & Gao, T. (2009) Oncogene, 28, 994–1004.
130) Newton, A.C. & Trotman, L.C. (2014) Annu. Rev. Pharmacol. Toxicol., 54, 537–558.
131) Smith, A.J., Wen, Y.A., Stevens, P.D., Liu, J., Wang, C., & Gao, T. (2016) Oncotarget, 7, 7801–7815.
132) Cross, D.A., Alessi, D.R., Cohen, P., Andjelkovich, M., & Hemmings, B.A. (1995) Nature, 378, 785–789.
133) Miura, T. & Tanno, M. (2010) Cardiovasc. Drugs Ther., 24, 255–263.
134) Pastorino, J.G., Hoek, J.B., & Shulga, N. (2005) Cancer Res., 65, 10545–10554.
135) Pantic, B., Trevisan, E., Citta, A., Rigobello, M.P., Marin, O., Bernardi, P., Salvatori, S., & Rasola, A. (2013) Cell Death Dis., 4, e858.
136) Cheung, E.C., Ludwig, R.L., & Vousden, K.H. (2012) Proc. Natl. Acad. Sci. USA, 109, 20491–20496.
137) Wolf, A.J., Reyes, C.N., Liang, W., Becker, C., Shimada, K., Wheeler, M.L., Cho, H.C., Popescu, N.I., Coggeshall, K.M., Arditi, M., & Underhill, D.M. (2016) Cell, 166, 624–636.
138) Marelli-Berg, F.M., Fu, H., & Mauro, C. (2012) Immunology, 136, 363–369.
139) Marko, A.J., Miller, R.A., Kelman, A., & Frauwirth, K.A. (2010) PLoS One, 5, e15425.
140) Palsson-McDermott, E.M. & O’Neill, L.A. (2013) BioEssays, 35, 965–973.
141) Gurel, E., Smeele, K.M., Eerbeek, O., Koeman, A., Demirci, C., Hollmann, M.W., & Zuurbier, C.J. (2009) J. Appl. Physiol., 106, 1909–1916.
142) Pasdois, P., Parker, J.E., & Halestrap, A.P. (2012) J. Am. Heart Assoc., 2, e005645.
143) De Duve, C. & Wattiaux, R. (1966) Annu. Rev. Physiol., 28, 435–492.
144) Levine, B. & Kroemer, G. (2008) Cell, 132, 27–42.
145) Yang, Z. & Klionsky, D.J. (2010) Nat. Cell Biol., 12, 814–822.
146) Ohsumi, Y. (2014) Cell Res., 24, 9–23.
147) Takeshige, K., Baba, M., Tsuboi, S., Noda, T., & Ohsumi, Y. (1992) J. Cell Biol., 119, 301–311.
148) Tsukada, M. & Ohsumi, Y. (1993) FEBS Lett., 333, 169–174.
149) Mizushima, N., Yoshimori, T., & Ohsumi, Y. (2011) Annu. Rev. Cell Dev. Biol., 27, 107–132.
150) Noda, T. & Ohsumi, Y. (1998) J. Biol. Chem., 273, 3963–3966.
151) Sengupta, S., Peterson, T.R., & Sabatini, D.M. (2010) Mol. Cell, 40, 310–322.
152) Wullschleger, S., Loewith, R., & Hall, M.N. (2006) Cell, 124, 471–484.
153) Yuan, H.X., Xiong, Y., & Guan, K.L. (2013) Mol. Cell, 49, 379–387.
154) Tan, V.P. & Miyamoto, S. (2016) J. Mol. Cell. Cardiol., 95, 31–41.
155) Ganley, I.G., Lam, H., Wang, J., Ding, X., Chen, S., & Jiang, X. (2009) J. Biol. Chem., 284, 12297–12305.
156) Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., Iemura, S., Natsume, T., Takehana, K., Yamada, N., Guan, J.L., Oshiro, N., & Mizushima, N. (2009) Mol. Biol. Cell, 20, 1981–1991.
157) Jung, C.H., Jun, C.B., Ro, S.H., Kim, Y.M., Otto, N.M., Cao, J., Kundu, M., & Kim, D.H. (2009) Mol. Biol. Cell, 20, 1992–2003.
158) Kim, J., Kundu, M., Viollet, B., & Guan, K.L. (2011) Nat. Cell Biol., 13, 132–141.
159) Nazio, F., Strappazzon, F., Antonioli, M., Bielli, P., Cianfanelli, V., Bordi, M., Gretzmeier, C., Dengjel, J., Piacentini, M., Fimia, G.M., & Cecconi, F. (2013) Nat. Cell Biol., 15, 406–416.
160) Russell, R.C., Tian, Y., Yuan, H., Park, H.W., Chang, Y.Y., Kim, J., Kim, H., Neufeld, T.P., Dillin, A., & Guan, K.L. (2013) Nat. Cell Biol., 15, 741–750.
161) Shang, L., Chen, S., Du, F., Li, S., Zhao, L., & Wang, X. (2011) Proc. Natl. Acad. Sci. USA, 108, 4788–4793.
162) Egan, D.F., Shackelford, D.B., Mihaylova, M.M., Gelino, S., Kohnz, R.A., Mair, W., Vasquez, D.S., Joshi, A., Gwinn, D.M., Taylor, R., Asara, J.M., Fitzpatrick, J., Dillin, A., Viollet, B., Kundu, M., Hansen, M., & Shaw, R.J. (2011) Science, 331, 456–461.
163) Gwinn, D.M., Shackelford, D.B., Egan, D.F., Mihaylova, M.M., Mery, A., Vasquez, D.S., Turk, B.E., & Shaw, R.J. (2008) Mol. Cell, 30, 214–226.
164) Inoki, K., Zhu, T., & Guan, K.L. (2003) Cell, 115, 577–590.
165) Takagi, H., Matsui, Y., Hirotani, S., Sakoda, H., Asano, T., & Sadoshima, J. (2007) Autophagy, 3, 405–407.
166) Bar-Peled, L., Schweitzer, L.D., Zoncu, R., & Sabatini, D.M. (2012) Cell, 150, 1196–1208.
167) Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A.L., Nada, S., & Sabatini, D.M. (2010) Cell, 141, 290–303.
168) Sancak, Y., Peterson, T.R., Shaul, Y.D., Lindquist, R.A., Thoreen, C.C., Bar-Peled, L., & Sabatini, D.M. (2008) Science, 320, 1496–1501.
169) Zoncu, R., Bar-Peled, L., Efeyan, A., Wang, S., Sancak, Y., & Sabatini, D.M. (2011) Science, 334, 678–683.
170) Duran, R.V., Oppliger, W., Robitaille, A.M., Heiserich, L., Skendaj, R., Gottlieb, E., & Hall, M.N. (2012) Mol. Cell, 47, 349–358.
171) Han, J.M., Jeong, S.J., Park, M.C., Kim, G., Kwon, N.H., Kim, H.K., Ha, S.H., Ryu, S.H., & Kim, S. (2012) Cell, 149, 410–424.
172) Hara, K., Yonezawa, K., Weng, Q.-P., Kozlowski, M.T., Belham, C., & Avruch, J. (1998) J. Biol. Chem., 273, 14484–14494.
173) Jewell, J.L., Kim, Y.C., Russell, R.C., Yu, F.X., Park, H.W., Plouffe, S.W., Tagliabracci, V.S., & Guan, K.L. (2015) Science, 347, 194–198.
174) Lorin, S., Tol, M.J., Bauvy, C., Strijland, A., Pous, C., Verhoeven, A.J., Codogno, P., & Meijer, A.J. (2013) Autophagy, 9, 850–860.
175) Roberts, D.J., Tan-Sah, V.P., Ding, E.Y., Smith, J.M., & Miyamoto, S. (2014) Mol. Cell, 53, 521–533.
176) Nojima, H., Tokunaga, C., Eguchi, S., Oshiro, N., Hidayat, S., Yoshino, K., Hara, K., Tanaka, N., Avruch, J., & Yonezawa, K. (2003) J. Biol. Chem., 278, 15461–15464.
177) Schalm, S.S. & Blenis, J. (2002) Curr. Biol., 12, 632–639.
178) Schalm, S.S., Fingar, D.C., Sabatini, D.M., & Blenis, J. (2003) Curr. Biol., 13, 797–806.
179) Tan, V.P. & Miyamoto, S. (2015) Autophagy, 11, 963–964.
180) Rambold, A.S., Cohen, S., & Lippincott-Schwartz, J. (2015) Dev. Cell, 32, 678–692.