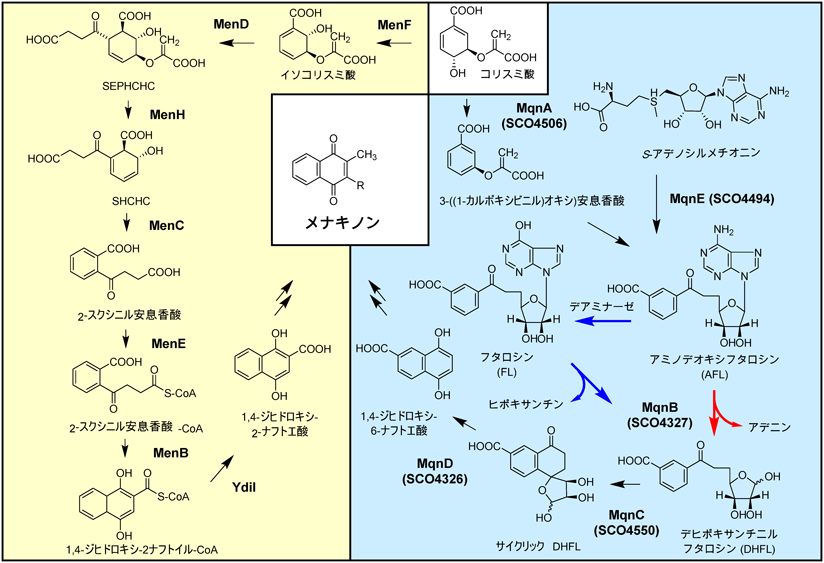
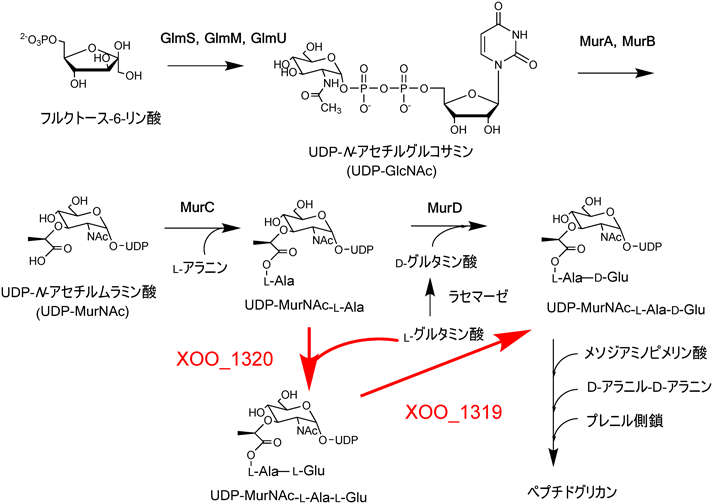
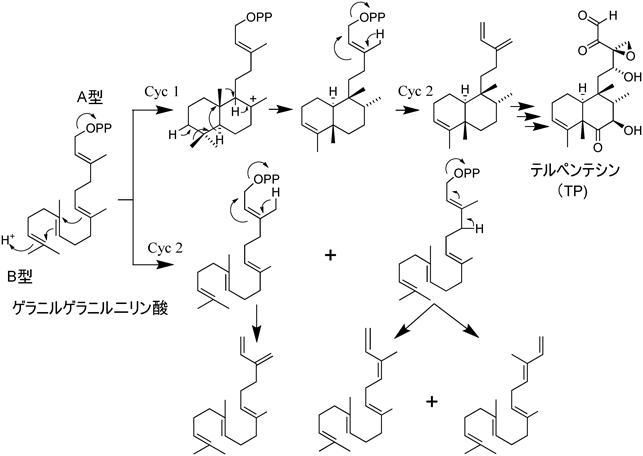
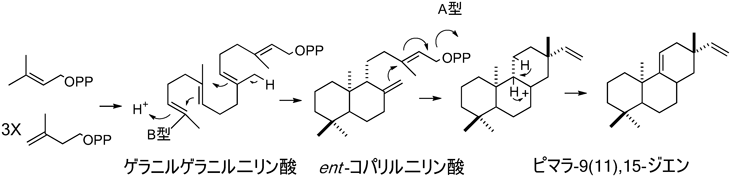
3) Bentley, R. & Meganathan, R. (1982) Microbiol. Rev., 46, 241–280.
37) Arnison, P.G., Bibb, M.J., Bierbaum, G., Bowers, A.A., Bugni, T.S., Bulaj, G., Camarero, J.A., Campopiano, D.J., Challis, G.L., Clardy, J., Cotter, P.D., Craik, D.J., Dawson, M., Dittmann, E., Donadio, S., Dorrestein, P.C., Entian, K.D., Fischbach, M.A., Garavelli, J.S., Göransson, U., Gruber, C.W., Haft, D.H., Hemscheidt, T.K., Hertweck, C., Hill, C., Horswill, A.R., Jaspars, M., Kelly, W.L., Klinman, J.P., Kuipers, O.P., Link, A.J., Liu, W., Marahiel, M.A., Mitchell, D.A., Moll, G.N., Moore, B.S., Müller, R., Nair, S.K., Nes, I.F., Norris, G.E., Olivera, B.M., Onaka, H., Patchett, M.L., Piel, J., Reaney, M.J., Rebuffat, S., Ross, R.P., Sahl, H.G., Schmidt, E.W., Selsted, M.E., Severinov, K., Shen, B., Sivonen, K., Smith, L., Stein, T., Süssmuth, R.D., Tagg, J.R., Tang, G.L., Truman, A.W., Vederas, J.C., Walsh, C.T., Walton, J.D., Wenzel, S.C., Willey, J.M., & van der Donk, W.A. (2013) Nat. Prod. Rep., 30, 108–160.
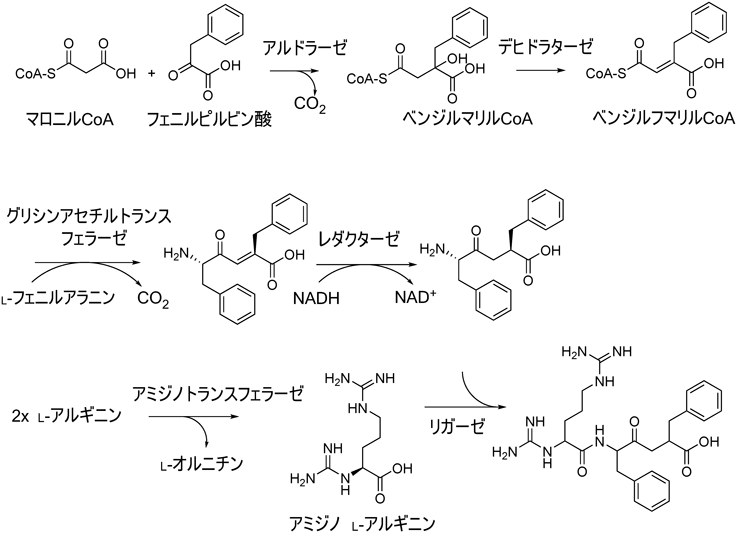
41) Noike, M., Matsui, T., Ooya, K., Sasaki, I., Ohtaki, S., Hamano, Y., Maruyama, C., Ishikawa, J., Satoh, Y., Ito, H., Morita, H., & Dairi, T. (2015) Nat. Chem. Biol., 11, 71–76.
45) Kawata, J., Naoe, D., Ogasawara, Y., & Dairi, T. (2017) Angew. Chem. Int. Ed. Engl., doi: 10.1002/anie.201611005