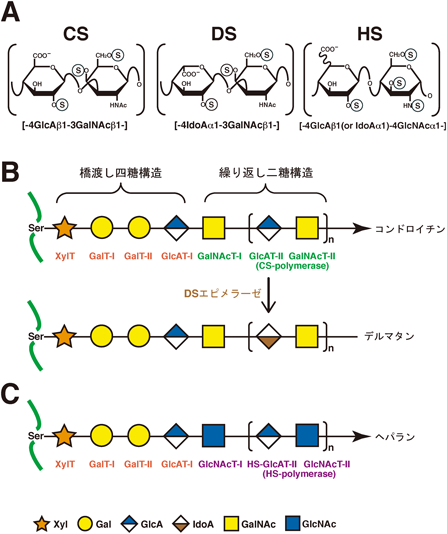
グリコサミノグリカン(glycosaminoglycan:GAG)の一種であるコンドロイチン硫酸(chondroitin sulfate:CS),デルマタン硫酸(dermatan sulfate:DS),ヘパラン硫酸(heparan sulfate:HS)は,ゴルジ体内でプロテオグリカン(proteoglycan:PG)のコアタンパク質の特定のセリン残基に付加され,細胞表面や細胞外マトリックスに輸送される.GAGは細胞接着,細胞のシグナル伝達の調節や細胞外マトリックスの構築など多様な生理機能を発揮する.CSは,グルクロン酸(glucuronic acid:GlcA)とN-アセチルガラクトサミン(N-acetylgalactosamine:GalNAc)の二糖単位が100~200回程度繰り返し重合した直鎖状の多糖鎖である.DSは,GlcAのC5位のカルボキシ基が異性化したイズロン酸(iduronic acid:IdoA)とGalNAcの二糖が重合した多糖鎖である(図1A).HSはGlcA(あるいはIdoA)とグルコサミン(glucosamine:GlcN)の二糖単位が重合した多糖鎖である(図1A).これらGAGは,構成する糖の各ヒドロキシ基やGlcNのアミノ基が硫酸化されることで多様性を獲得し,さまざまなタンパク質と相互作用し種々の機能を発揮するようになる.
CS, DS, HSは,コアタンパク質上の特定のセリン残基に,共通の橋渡し四糖構造,キシロース-ガラクトース-ガラクトース-グルクロン酸(Xyl-Gal-Gal-GlcA)を介して共有結合している(図1B).この橋渡し四糖の合成に関わる糖転移酵素をコードする遺伝子の変異により,骨や皮膚,結合組織,心臓の形成不全を示す一群の遺伝性疾患「プロテオグリカンリンカー病(proteoglycan linkeropathy:PG-linkeropathy)」が発症する.PG-linkeropathyは,GAGの橋渡し四糖構造の生合成に関わる糖転移酵素の変異によって発症する多様な結合組織疾患の総称で,理化学研究所の池川志郎博士によって名づけられた1).本稿では,PG-linkeropathyについて紹介する.
GAGの橋渡し四糖構造の合成は,キシロース転移酵素(XylT)によってウリジン二リン酸(UDP)-Xylからコアタンパク質のセリン残基にXylを転移することにより開始される(図1B).次いで,UDP-Galからβ4ガラクトース転移酵素-I(GalT-I)およびβ3ガラクトース転移酵素-II(GalT-II)によって,UDP-GlcAからβ3グルクロン酸転移酵素-I(GlcAT-I)によって順次各糖残基が付加され,四糖構造が合成される(図1B, C).
橋渡し四糖構造の末端のGlcA残基に,CS合成酵素群がUDP-GalNAcからGalNAcを転移するとCSの二糖繰り返し領域[GlcA-GalNAc]nが合成される(図1B).一方HSポリメラーゼによりGlcNAcが転移されると,HSの二糖繰り返し領域[GlcA-GlcNAc]nが合成される(図1C).また,DSの二糖繰り返し領域[IdoA-GalNAc]nは,前駆体であるコンドロイチン骨格[GlcA-GalNAc]nが合成された後,もしくは伸長反応の途中で,DSエピメラーゼがGlcA残基のC5位のカルボキシ基を異性化することにより形成される(図1B).CS, DS, HSのそれぞれの基本骨格が生合成された後,種々の硫酸基転移酵素が各糖残基のヒドロキシ基やGlcNのアミノ基に硫酸基を転移し,成熟したGAGになる.
3.GAGの橋渡し領域四糖の生合成不全によるPG-linkeropathy(表1)
表1 GAGの橋渡し領域四糖の生合成に関与するヒトの糖転移酵素とPG-linkeropathy| タンパク質・酵素活性 | 遺伝子名 | MIM*ナンバー | ヒトの遺伝病 |
|---|
| キシロース転移酵素(XylT) | XYLT1 | 608124 | 2型デビュクオア骨異形性症 |
| 615777 |
| XYLT2 | 605822 | 骨脆弱性,白内障,難聴を伴う脊椎・眼異形成症 |
| 608125 |
| β4ガラクトース転移酵素-I (GalT-I) | B4GALT7 | 130070 | エーラス・ダンロス症候群(早老性I型),レユニオン島型ラーセン症候群 |
| 604327 |
| β3ガラクトース転移酵素-II (GalT-II) | B3GALT6 | 271640 | エーラス・ダンロス症候群(早老性II型),関節弛緩を伴う脊椎骨端骨幹端異形成症I型 |
| 615291 |
| 615349 |
| β3グルクロン酸転移酵素-I (GlcAT-I) | B3GAT3 | 245600 | ラーセン症候群,低身長 |
| 606374 |
| * Mendelian Inheritance in Man. |
1)XYLT1の変異による2型デビュクオア骨異形性症
橋渡し四糖構造の最初の糖であるXyl残基の転移に関わるXYLT1のホモ変異によって骨系統疾患が発症する2).一塩基置換によるアミノ酸変異(p.Arg481Trp)を持つ患者は,低身長,特徴的顔貌,脂肪分布の異常,中程度の知的障害を示す.患者由来の細胞のXYLT1は一部がゴルジ体に局在するものの,大部分が細胞質に分布していた.また患者由来の細胞のデコリン-PGは,糖鎖を欠損していた.
XYLT1の別のホモ変異p.Arg598Cys, p.Arg147*, p.Pro93Alafs*69(fs*69はフレームシフトによって,アラニンから69番目のリーディングフレームが終止コドンになることを表す)と2か所のイントロン内のホモ変異(c.1290–2A>C, c.1588–3C>T)のいずれかの変異によって,2型デビュクオア(Desbuquois)骨異形成症を発症する3).デビュクオア骨異形成症は,低身長症,重篤な生後発達遅延,眼の突出を伴う平らな顔,大関節の脱臼,関節弛緩症を主症状とする常染色体劣性の遺伝病である.患者由来の線維芽細胞では,HSよりむしろCSの合成が低下していた.XYLT1はCS-PGのコアタンパク質のセリン残基にXylを優先的に転移するのかもしれない.また,前述のp.Arg481Trpの変異を有する患者と比較して,2型デビュクオア骨異形性症患者は,より重篤な骨の形成異常を示した2, 3).その理由としては,p.Arg481Trpの変異はXYLT1の部分的な機能低下で,GAGの合成量や症状に対する影響もより部分的なので,その変異による患者の症状が比較的軽いのではないかと考えられている.今後,両患者が産生するGAG鎖の詳細な解析によって,症状の違いが説明できるかもしれない.
2)XYLT2の変異による脊椎・眼症候群
XYLT1のアイソフォームであるXYLT2のホモ変異(p.Ala174Profs*35)によって,脊椎・眼症候群が発症する4).本患者は,学習困難,網膜剥離,弱視,眼振,聴覚障害,心臓の中隔欠損,骨の脆弱性などの症状を示す.生化学的解析により,患者由来の皮膚線維芽細胞では健常人由来の細胞と比較して,キシロース転移酵素活性が低下しており,GAGの生合成量も減少していた.TaylanらはXYLT2の別のホモ変異(p.Arg730*, p.Arg563Gly, p.Leu605Pro)による脊椎・眼症候群を報告している5).ナンセンス変異(p.Arg730*)を有する患者は,p.Ala174Profs*35変異を示す患者と症状が似ているが,p.Arg563Glyおよびp.Leu605Proの変異を有する患者はより症状が軽い.前二者はXYLT1の機能完全欠失型,後二者は部分欠失型であると考えられる.
以上の知見から,XYLT1とXYLT2は互いの機能を補うことはできないことが明らかとなった.また,これら遺伝子に変異を持つ患者間では骨や眼の症状が違うことから,XYLT1とXYLT2ではXylを付加するPGのコアタンパク質に違いがあると考えられる.
3)B4GALT7/GalT-Iの変異による早老性型エーラス・ダンロス症候群とレユニオン島型ラーセン症候群
エーラス・ダンロス症候群(Ehlers-Danlos syndrome:EDS)は,皮膚の伸展性,関節弛緩,血管脆弱性などを特徴とする結合組織疾患の総称である.1998年の改訂国際分類では古典型,関節過動型,血管型,後側彎型,多発関節弛緩型,皮膚脆弱型の六つに大別された.これらの多くは,コラーゲンあるいはその修飾酵素の遺伝子変異によって引き起こされることが生化学的・遺伝学的解析から明らかとなっていた.しかし,近年従来とは異なる新しいタイプのEDSが次々に報告されている.その一部がDSの合成不全によるタイプである.
橋渡し四糖構造の2番目のGal残基を1番目のXyl残基に転移するGalT-IをコードするB4GALT7の複合ヘテロ接合変異(p.Leu206Proとp.Ala180Asp)やホモ変異(p.Arg270Cys)によって,早老性型EDSが発症する6–8).患者は早老様顔貌,低身長症,全身性骨粗鬆症,創傷治癒の遅延,関節の過伸展,弾性があり弛緩した皮膚,低緊張性筋肉などの多様な症状を示す6).当初1990年代の研究によって,DS鎖の欠損と考えられていたが,最近の知見から,CSやHSも部分的に短い鎖が合成されていることがわかった9, 10).したがって,早老性型EDSの多様な症状は,DSだけでなく,CSやHSも部分的に欠損しているためかもしれない.
また,レユニオン島型ラーセン症候群(Larsen syndrome in Reunion Island)を発症する患者においても,B4GALT7のホモ変異(p.Arg270Cys)が見つかった11).ラーセン症候群は,頭蓋顔面異形成,多発性の関節脱臼,内反尖足を特徴とする骨系統疾患である.レユニオン島型ラーセン症候群患者では,従来のラーセン症候群の症状に加えて,低身長症,特徴的な顔貌,関節弛緩を示し,早老性型EDSと重複する部分が多いと報告されている11).同一遺伝子の同じ変異にも関わらず,EDSとラーセン症候群の二つの疾患が発症する理由は不明である.今後,詳細に解析する必要がある.
4)B3GALT6/GalT-IIの変異による関節弛緩を伴う脊椎骨端骨幹端異形成症とエーラス・ダンロス症候群
橋渡し四糖構造の3番目のGal残基の転移に関わるGalT-IIをコードするB3GALT6遺伝子の変異によって,関節弛緩を伴う脊椎骨端骨幹端異形成症I型(spondyloepimetaphyseal dysplasia with joint laxity type 1:SEMD-JL1)を発症する1).SEMD-JL1は,脊柱の変形や関節の脱臼などの重度の骨格異常を起こす骨系統疾患である.全エキソーム解析の手法により,7家系の患者の遺伝子変異を調べたところ,患者はすべてB3GALT6遺伝子の複合ヘテロ接合性変異(p.Met1?, p.Ser65Gly, p.Pro67Leu, p.Asp156Asn, p.Arg232Cys, p.Cys300Ser)を有していた.野生型の組換え型GalT-IIと比較して,各変異型の組換えタンパク質のGal転移活性は有意に低下していた.また驚いたことに,早老性型EDS患者においても,SEMD-JL1と同様にB3GALT6遺伝子の複合ヘテロ接合性変異(p.Arg6Tryp, p.Asp118Alafs*160, p.Met139Ala141del, p.Arg197Alafs*81, p.Ser309Thr)を有していることがわかった1).さらに,MalfaitらはEDS患者で別のホモ変異(p.Asp207His, p.Gly217Ser)と複合ヘテロ接合変異(p.Ala108Glyfs*163, p.Asp207His)を見いだしている12).早老性型EDSとSEMD-JL1とでは,B3GALT6遺伝子の変異部位が異なっているので,GalT-IIの機能低下の程度が異なり,合成されるCS, DS, HSの量や長さが異なることで,症状に違いが生じる可能性が考えられる.実際Malfaitらは患者由来の線維芽細胞におけるCS, DSおよびHS鎖の合成量の低下を示している12).
5)B3GAT3/GlcAT-Iの変異によるラーセン症候群と低身長・脊柱側彎症,多発性骨折・骨粗鬆症
橋渡し四糖構造のGlcAを付加するGlcAT-IをコードするB3GAT3遺伝子のホモ変異(p.Arg277Gln)によって,心臓弁の形成不全を伴うラーセン症候群を発症する家系がアラブ首長国連邦で見いだされた13).B3GAT3変異患者の皮膚の線維芽細胞では,顕著にGlcA転移活性が低下し,CS, DS, HS鎖の合成量も低下していた13).
別のGlcAT-Iのホモ変異(p.Pro140Leu)によって,脊柱側彎症,低身長などの骨格異常を呈する患者がインドネシアのニアス島で発見された14).しかしながら,上述したp.Arg277Gln変異患者でみられた心臓弁の形成不全はなかった.患者由来のリンパ芽球細胞においては,GlcA転移酵素活性が有意に低下しCS, DS, HS鎖の合成量が顕著に低下していた.
GlcAT-Iの複合ヘテロ接合変異(p.Met1?, p.Leu224Gln)によって,重篤な骨粗鬆症,筋緊張低下,心形成異常,骨格異常を来す患者が見いだされた15).p.Leu224Gln変異型のGlcAT-Iは野生型のGlcAT-Iと比較して,糖転移活性が顕著に低下していた.また,この患者ではGlcAT-IをコードするB3GAT3遺伝子の開始コドンのアデニンがグアニンに変異していた.解析の結果,GlcAT-Iの開始コドンとは別のATGから翻訳されていることが示唆された.この変異型のタンパク質は,読み枠がずれており,糖転移酵素に特徴的なモチーフを保持していないことから,完全欠失型の変異であると考えられる.さらに,健常人由来の線維芽細胞と比較して,CSとDSの合成量が顕著に低下していた.
以上の三つの研究から,同じGlcAT-Iの変異でも,その変異部位の違いによって,骨格や心臓の形成異常の症状の多様性が生じ,異なった疾患として認識される表現型となったと考えられる.今後,三つの患者由来の細胞のGlcAT-I活性やPGとGAG量を比較して検討する必要があろう.
GAGに共通の橋渡し領域四糖の生合成に関わる糖転移酵素の変異によって,さまざまな骨,皮膚,結合組織異常を来す遺伝性疾患が発症することが明らかとなった.しかしながら,その合成を担う糖転移酵素(XYLT1, XYLT2, B4GALT7, B3GALT6, B3GAT3)は,共通のGAGの生合成を担っているにも関わらず,それぞれの酵素遺伝子の変異により異なった症状を示すこと,さらに同一遺伝子の変異でも変異部位の違いで症状が異なることの理由は不明である.おそらく,変異部位の違いによって各糖転移酵素の機能低下の程度に違いが生じ,生合成されるGAGの長さや量,硫酸化の程度など,GAGの微細構造・質が各患者で異なっているのであろう.各患者で合成されるGAGの長さや量,影響を受けるPGのコアタンパク質などを解析することは,発症メカニズムの解明に有効だろう.また,β4GalT, β3GalT, β3GlcATの各糖転移酵素ファミリーの他の相同タンパク質が,GAGの結合領域の糖転移酵素の機能を部分的に補っているのかもしれない.橋渡し領域四糖のXyl残基とGal残基はそれぞれリン酸化や硫酸化修飾を受け,GAG生合成の制御を担っていると考えられていることから,これらの修飾構造の影響も調べる必要がある.
今後,橋渡し領域四糖の生合成に関わる糖転移酵素やその反応産物である四糖構造によるGAG生合成の制御・調節機構,GAGと結合し,軟骨や皮膚の発生に関わるタンパク質の解析,GAGの機能ドメイン,シグナル伝達経路などの研究によって,PG-linkeropathyの治療法や治療薬が開発されることが期待される.