1. 血友病A治療における課題に対する抗体医薬応用の発想
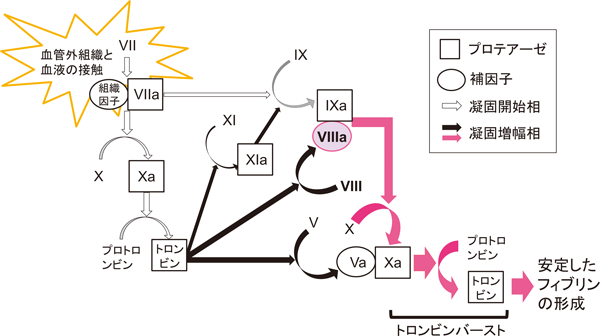
血友病Aは,血液凝固第VIII因子(FVIII)の先天性欠乏に起因する出血性の疾患であり,FVIIIの活性が正常の1%未満である重症例,および1~5%の中等症例の一部では,小児期より重篤な出血を繰り返す1).FVIIIは,凝固反応の初期段階(凝固開始相)で生じる少量のトロンビンあるいは活性型第X因子(FXa)により活性化されると補因子機能を呈し(活性型FVIII;FVIIIa),活性型第IX因子(FIXa)が触媒する第X因子(FX)活性化反応を飛躍的に促進する役割を有する.その結果生じたFXaは,その補因子である活性型第V因子(FVa)とともに,プロトロンビンをトロンビンへと爆発的に変換する(図1).これはトロンビンバーストと呼ばれ,大量に生じたトロンビンは,フィブリノゲンを凝固反応の最終産物であるフィブリンに変換する.血友病Aでは,FIXa触媒FX活性化反応の補因子であるFVIIIが欠乏しているためFXaが十分生じず,トロンビンバーストが不完全となり,その結果,止血栓におけるフィブリン形成が不足する.そのため,重篤な出血傾向を呈する.
血友病Aの標準治療として,数十年来,FVIII製剤による補充療法が実施されている.近年は,出血が生じたときに止血するために使用されるオンデマンド止血療法のみならず,出血を予防するため,長期間にわたって定期的にFVIII製剤を補充する定期補充療法が広まってきた.定期補充はFVIII活性を常に1%以上に保つこと,すなわち,中等症(FVIII活性:1~5%)以上のレベルに保つことを目標に実施される1, 2).これにより,出血回数を大きく減らすのみならず,乳幼児期から早期に定期補充を継続すれば,関節出血の反復に起因する血友病性関節症の進展を阻止することができる1, 3).しかしながら,通常のFVIII製剤の半減期は約半日,2015年に上市された半減期延長型FVIII製剤の半減期でも約0.75日といずれも短く,また皮下からの吸収性が非常に低いため,出血予防のための定期補充の実施には,週に1~3.5回の静脈注射が必要となる.特に乳幼児期においては細い静脈に注射を実施する困難さもあり,患者およびご家族の大きな負担となるばかりか,定期補充の早期導入のハードルにもなっている1).また,FVIII製剤は,FVIII欠損症例や変異症例において外来抗原となるため,約30%の重症患者にFVIIIに対する同種抗体(FVIIIインヒビター)が発生する1).FVIIIインヒビターが発生すると標準治療薬のFVIII製剤の効果が激減または消失するため,止血コントロールに難渋することが多く,患者のQOL(生活の質)は大きく低下する1).
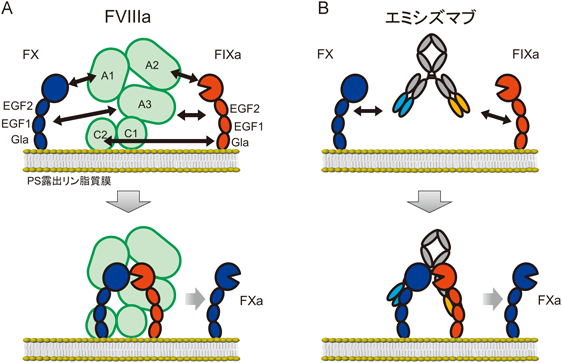
現代の血友病A治療における課題を克服する新薬の創出にあたり,我々は抗体医薬に注目した.抗体は,FVIIIと同様に高分子であり,一般的特性として,長い血中半減期および高い皮下吸収性を有する.そこで,もし抗体をもってFVIII補因子作用を発揮できれば,長期持続型の皮下投与製剤となりうる上,FVIIIとは抗原性が異なることからFVIIIインヒビター存在の有無に関わらず効果を発揮することも期待できる.すなわち血友病Aの標準治療法におけるアンメットニーズに十分に応えることができるのではないかと考えた.折しも,FIXaおよびFXに対するFVIIIaの結合様式を解明した論文が相次いで報告されたことを基に4–6),我々は「FVIIIaは,活性化血小板上などホスファチジルセリン(PS)が露出したリン脂質膜表面にて,FIXaのプロテアーゼ活性中心をFXの切断部位に正確に配置させることで補因子活性を示す」という仮説を立て(図2A),抗体をもってこの補因子作用を実現するために,FIXaおよびFXに対するバイスペシフィックIgG抗体を用いるという発想に至った(図2B)7).公知の構造解析データを調べると,FVIIIa分子内のFIXa結合部位とFX結合部位との間の距離と,IgG分子内の二つの抗原認識部位間の距離は同じ程度であり,本発想に挑戦する価値があると考えた.
2. FVIII補因子作用を有するバイスペシフィック抗体“エミシズマブ”の創製
しかしながら,目指す作用を実現させるためには,PS露出リン脂質膜表面においてバイスペシフィック抗体がFIXaのプロテアーゼ活性中心をFXの切断部位に精緻に配向させる必要がある.したがって,目的のバイスペシフィック抗体の創出は容易ではないと考えられた.そこで,我々は,万単位のバイスペシフィック抗体を作製し,そこからプロトタイプのバイスペシフィック抗体を見いだす方策を立てた7, 8).まず,配列の多様性を得るために複数の動物種(マウス,ラットおよびウサギ)にFIXaまたはFXを免疫し,各抗原に対する抗体を約200ずつ取得した.次いで,これら抗体の可変領域をクローニングし,それらを組み合わせて,ヒトIgGキメラ抗体として,約4万の抗FIXa/FXバイスペシフィック抗体をHEK細胞にて一過性に発現させた.約4万のバイスペシフィック抗体の個々について,純化凝固因子を用いたFIXa触媒FX活性化反応アッセイにてスクリーニングを行い,同反応を促進する補因子活性を基に,数百のバイスペシフィック抗体を選抜した.さらに,FVIII欠乏血漿における内因系凝固時間短縮能等を基準に絞り込みを進め,プロトタイプのバイスペシフィック抗体BS15を同定した.しかしながら,BS15の補因子活性は,臨床応用するには十分とはいえなかった.また,目的とする医薬品として完成させるためには,薬理活性(補因子活性)の向上や抗体のヒト化のみならず,物理化学的安定性の向上(→製剤の保存安定性),溶解性の向上(→製剤の抗体濃度:皮下投与のためには高濃度化が必要),非特異結合の低下(→皮下吸収性),in silico免疫原性予測値の改善(→免疫原性)等,多様な要素につき改良を行う必要があった.我々は,アミノ酸置換によってバイスペシフィック抗体分子を改変し(バリアント),これら要素の改良を進めた.具体的には,一度に数十のバイスペシフィック抗体バリアントをデザインして作製し,個々のバリアントにつき各要素の評価をし,有望と思われるアミノ酸置換を見いだしていった.有望視されたアミノ酸置換であってもすべての要素を必ずしも改善するものではないため,当該アミノ酸置換が逆に改悪につながる要素については別の箇所のアミノ酸置換で改善して補完することを考えながら,次の数十のバリアントをデザインした.このサイクルを繰り返し,約2400ものバイスペシフィック抗体バリアントをデザイン・評価した末に,最終的に,補因子活性に加え他の要素も優れたヒト化バイスペシフィック抗体“ACE910(一般名:エミシズマブ)”を同定した8).
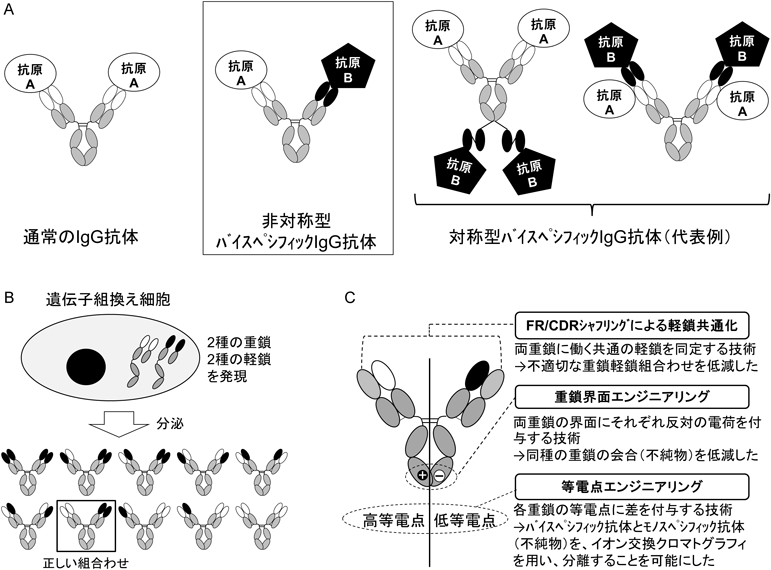
一方で,我々は,抗FIXa/FXバイスペシフィック抗体を医薬品として開発するために,工業製造の課題を解決する必要もあった.本バイスペシフィック抗体は,非対称型バイスペシフィックIgG抗体の形態を有する(図3A).この非対称型バイスペシフィックIgG抗体をそのまま遺伝子組換え細胞内で発現させると,2種類の重鎖と2種類の軽鎖がランダムに組み合わされ,所望の組合わせの他に9種類もの不純抗体が形成される(図3B).不純抗体の形成は,所望のバイスペシフィック抗体の発現量を低下させるのみにとどまらない.形成された10種の抗体間の特性の類似性から,抗体医薬製造に用いられる通常の手法では不純抗体の分離除去が困難であることも,工業製造上の大きな問題となる.そこで,通常のIgG抗体と同様の効率と品質の工業製造を可能とすべく,我々は,新たに3種の抗体工学技術(図3C)を開発し,エミシズマブに適用した8).なお,エミシズマブの創製過程においては,プロトタイプ抗体選抜の最終段階で,3種の技術の一つFR/CDRシャフリングによる軽鎖共通化の適用可能性も評価し,選抜されたBS15は,共通軽鎖を有するプロトタイプ・バイスペシフィック抗体として同定された8).
前述のとおり,我々は,「FVIIIaは,PSが露出したリン脂質膜表面にて,FIXaのプロテアーゼ活性中心をFXの切断部位に正確に配置させることで補因子活性を示す」という仮説の下,エミシズマブを創製した.エミシズマブが同仮説に基づいて作用していることを,純化凝固因子を用いたFIXa触媒FX活性化アッセイで証明した.具体的には,FIXaまたはPS露出リン脂質膜が存在しない場合はエミシズマブがFX活性化反応を促進しないことを示して,エミシズマブがPS露出リン脂質膜依存性の補因子であることを証明した9).さらに,エミシズマブの片側の腕からなる抗FIXa抗体および抗FX抗体を同時に存在させてもFX活性化反応を促進しないことを示して,バイスペシフィック抗体として両抗原を架橋して初めて同反応を促進することを証明した8).エミシズマブ自体は,FVIIIaと異なり,PS露出リン脂質膜に直接結合しない.それにも関わらずエミシズマブにPS露出リン脂質膜依存性があるのは,エミシズマブ単独では,FIXaとFXの位置を精緻に合わせられないことに起因すると考察された.すなわち,エミシズマブ単独では1か所のみの架橋にとどまり,FIXaのプロテアーゼ活性中心をFXの切断部位に精緻に配置するには自由度が大きすぎるが,エミシズマブおよびPS露出リン脂質膜の両者が存在すれば,2か所で架橋して両因子をしっかり固定することが可能となり,FIXaのプロテアーゼ活性中心をFXの切断部位に精緻に配置できるのだろうと考察した.このPS露出リン脂質膜依存性は,エミシズマブが,FVIII(a)と同様,止血・血栓部位特異的に働くことを示している.
さらに,エミシズマブの補因子活性を,反応速度論に基づき定量的に解析した9).FIXa触媒FX活性化反応において,エミシズマブは,補因子のない状態に比し,kcatを4480倍,Kmを19.5倍改善し,酵素反応効率kcat/Kmを87,400倍改善した.FVIIIaとの比較では,kcatの改善率は1/43.8倍,Kmの改善率は3.86倍,kcat/Kmの改善率は1/11.3倍という値であった.エミシズマブのkcat/Kmの改善率は,正常の1%のFVIIIが存在すれば臨床上意義のある活性が示されることを考慮すると,有望なものと考えられた.
なお,凝固のネガティブフィードバック因子のアンチトロンビンがFIXaおよびFXaに,またtissue factor pathway inhibitor(TFPI)がFXaに作用することから,これらに対するエミシズマブの拮抗作用についても検討を実施した.その結果,エミシズマブはアンチトロンビンおよびTFPIの作用を阻害することはなく,すなわちこれら因子がエミシズマブの活性に無関係であることが示された10).
また,エミシズマブはFVIIIaと異なり活性化プロテインC(APC)による不活化を受けないことも考慮すべき点である.この点について,我々は,in vitro検討(血漿トロンビン生成アッセイ)を実施し,エミシズマブ存在下であっても,APCによるFVaの不活化を通じて,APCによる凝固のネガティブフィードバックが十分働くことを示した11).
4. 血漿におけるエミシズマブのin vitro活性
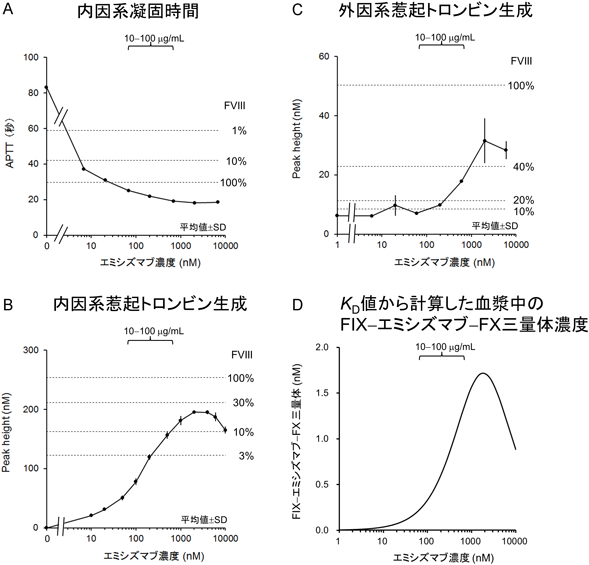
エミシズマブの補因子作用は,血漿を用いたin vitroアッセイでも確認された.in vitroにてFVIII欠乏血漿にエミシズマブを添加すると,FVIIIを添加した場合と同様,内因系凝固の一指標である活性化部分トロンボプラスチン時間(activated partial thromboplastin time:APTT)が短縮した(図4A).このAPTTの短縮は第I相臨床試験においても確認された12, 13).なお,エミシズマブ濃度によっては,FVIII 100%におけるAPTT値より短い値を示したが,この解釈には注意を要する.FVIIIの活性発現に際しては,血漿凝固反応の最初に生じる少量のトロンビンやFXaによって活性化を受ける必要があり,そのための時間もAPTTに加算される.一方,エミシズマブは活性化を要さず補因子活性を発揮するため,FVIIIと異なり活性化のための時間がAPTTに加算されることはない.したがって,エミシズマブ存在下では,APTT値による従来の判断基準が必ずしも適用できない.そこで,我々は,血漿におけるエミシズマブのin vitro活性評価の一環として,血漿トロンビン生成アッセイも実施した.同アッセイのパラメータの一つであるPeak heightは,トロンビンバースト(図1)の大きさを反映する.その結果,FVIII欠乏血漿に添加したエミシズマブは,同パラメータにおいて,最大でFVIII 10%以上に匹敵する活性を有することが示された(図4B, C).
エミシズマブの抗原に対する結合活性は表面プラズモン共鳴にて解析した.エミシズマブは,一方の腕でFIX/FIXaのEGF様領域-1,他方の腕でFX/FXaのEGF様領域-2に結合し(図2B),それぞれに対する結合解離定数(KD値)は,1.58 μM/1.52 μM, 1.85 μM/0.978 μMであった.これらKD値は,他の抗体医薬のKD値(1桁nMかそれ以下)に比し格段に大きく,すなわち結合が比較的弱いことが示された9).凝固カスケードにおいては,反応の結果生じたFXaは補因子から離れ,次の反応に供される必要があり,FXaに対する結合活性が強くないことは理に適っている.
このKD値を用いて,循環血漿中の抗原抗体複合体形成量を計算したところ,FIX, FX,エミシズマブのごく一部のみがFIX–エミシズマブ–FX三量体を形成していると予測された9).このFIX–エミシズマブ–FX三量体濃度は,エミシズマブ濃度に対して比例の関係になく,エミシズマブ濃度約265 µg/mL地点をピークとする釣鐘状の濃度反応性を示すと予測された(図4D)9).このベルシェープの濃度反応性は,血漿アッセイでも再現されており(図4A~C),血漿中のFIX–エミシズマブ–FX三量体濃度が,止血・血栓の場で形成される酵素(FIXa)–補因子(エミシズマブ)–基質(FX)複合体の量,すなわちエミシズマブの補因子活性に相関していることが示唆された9).
また,KD値を用いた計算により,国内第I相臨床試験(後述)で薬効を示した血漿エミシズマブ濃度範囲(10~100 µg/mL)においては,循環FIXおよびFXの大半が単体で存在すると推測された9).すなわち,理論上,凝固カスケードの他の反応に対するエミシズマブの干渉が,あったとしても小さいことを示しており,これは治療薬として望ましい特性と考えられる.
なお,エミシズマブは,前述のとおりAPTT短縮作用が強いため,APTTをベースとした臨床検査(たとえば,FVIII活性,FVIIIインヒビター力価,FIX活性,プロテインC活性)に強く干渉する.したがって,エミシズマブ存在下でこれらを測定する場合には工夫が必要であり,現在までに,抗エミシズマブ抗体をin vitroで血漿試料に添加しエミシズマブの作用を中和する方法が提唱されている14).また,FVIIIインヒビター力価測定に関しては,エミシズマブが作用しないウシ因子で構成される既存の市販キットを用いるアイデアも一手法としてあげられる.
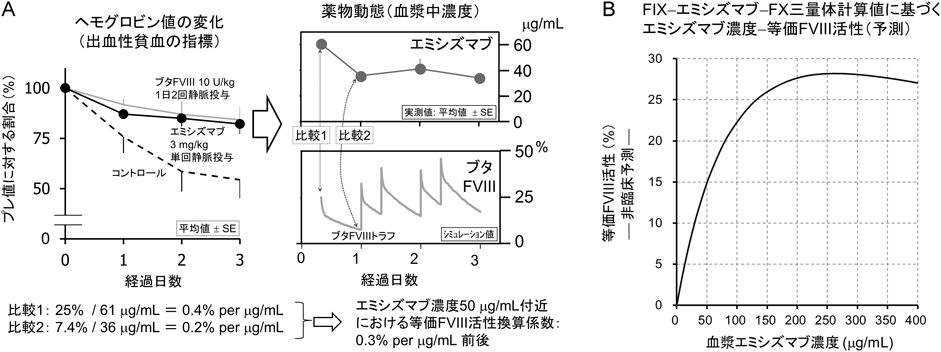
in vitro血漿アッセイでの効果がin vivoの止血作用につながるか否かを,エミシズマブ作用の動物種特異性を鑑み,カニクイザルを用い検証した.まず,抗FVIII中和抗体を作製し,同抗体を投与することで後天性血友病Aモデルを確立した.この際,陽性対照として遺伝子組換えブタFVIIIを用いる目的で,抗FVIII中和抗体としては,ブタFVIIIには交叉しない抗体をあらかじめ準備した.同後天性血友病Aカニクイザルにて人工的に惹起した出血に対して,エミシズマブ3 mg/kg単回静脈内投与は,遺伝子組換えブタFVIII 10 U/kg 1日2回静脈内投与と同程度の止血効果を示した15).この結果を基に,両被験物質の血中動態の違いを考慮して,エミシズマブの等価FVIII止血活性の推定を試みた.エミシズマブに若干有利な投与直後の比較では61 µg/mLのエミシズマブが25%のFVIIIに相当し,エミシズマブに若干不利なFVIIIトラフ(最低値)での比較では36 µg/mLのエミシズマブが7.4%のFVIIIに相当すると示唆された(図5A).すなわち,本カニクイザルモデルにおいて,血漿エミシズマブ濃度50 µg/mL前後ではおおまかに1 µg/mLあたりブタFVIII 0.2~0.4%(約0.3%)に相当する止血活性が発揮されたと推測された.血漿FIX–エミシズマブ–FX三量体濃度がエミシズマブの補因子活性に相関するという前述の仮説と合わせて適用すると,非臨床における血漿エミシズマブ濃度と等価FVIII活性の関係は,図5Bのように推測された.さらに,カニクイザルにて後天性血友病A状態を8週間維持したところ,対照群では全例に関節出血に伴う運動障害がみられたのに対し,エミシズマブ投与群(初回3.97 mg/kg +週1回1 mg/kg皮下投与;血漿エミシズマブ濃度20~50 µg/mL)では,それが完全に抑制された16).
Den Uijlらは,血友病A患者において,生来のFVIII残存活性が3%以上存在すると関節出血回数が大きく減り,12%以上存在すると関節出血回数がほぼゼロになるという疫学的解析結果を報告したが17),上記動物実験で予測されたエミシズマブの等価FVIII活性は,この報告と矛盾がない.
また,同じくカニクイザルを用い,エミシズマブの薬物動態を検討した15).エミシズマブ単回投与試験において,皮下投与の血中半減期(β相半減期)は19.4~26.5日であり,皮下投与時の生物学的利用率はほぼ100%であった.
国内第I相臨床試験は,健康成人(パートA, B)および重症血友病A患者(パートC)を対象として,2012年に開始された.健康成人パートは,単回皮下投与・プラセボ対照・ランダム化・二重盲検・個体間用量漸増試験であった(最高用量:1 mg/kg)12).本パートにおいて,1 mg/kgまでのエミシズマブ単回皮下投与の安全性・忍容性が確認され,薬物動態に関しては,カニクイザル試験と同様,長い血中半減期(β相半減期:28.3~34.4日)と良好な皮下吸収性が示された.
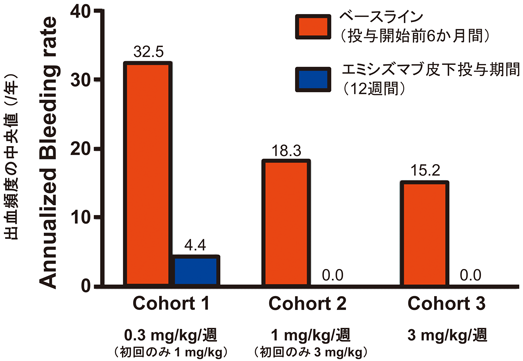
患者パートは,FVIIIインヒビター保有および非保有の重症血友病A患者を対象とした,反復皮下投与(12週間)・個体間用量漸増試験(3群,各群6例)であった13).出血予防を期待できるレベルとして,前述の非臨床試験による予測値に基づき,血漿エミシズマブ維持濃度を10, 33, 100 µg/mL前後に設定した.33 µg/mLでの等価FVIII活性期待値はおおむね10%に当たる.週1回皮下投与で上記目標濃度を達成できるエミシズマブ投与量を,カニクイザルを用いた血中動態試験データを基に計算し,各群の用量を0.3 mg/kg(初回のみ1 mg/kg),1 mg/kg(初回のみ3 mg/kg),3 mg/kgに設定した.
なお,エミシズマブ製剤の抗体濃度は最大約150 mg/mLであり,この場合,たとえば,1 mg/kgの用量では,体重75 kgの患者に対し約0.5 mLの投与ボリュームとなる(なお第I相臨床試験の製剤は約80 mg/mLであった).理論上,用量を増量すれば投与間隔を延ばすことができるが,皮下投与ボリュームが増大し,投与ごとの負担は増えることとなる.
本試験の結果,エミシズマブ投与により,投与開始前に比し出血頻度の著明な減少が認められた(図6).特に,1および3 mg/kg/週の2群においては,出血頻度の中央値がゼロであった.前述のとおり,非臨床試験による予測ではこれら群においておおむね10%以上の等価FVIII活性が期待されていた.したがって,前述のDen Uijlらの疫学的解析結果17)と照らし合わせると,期待に矛盾しない結果であった.
本試験において,血栓性の有害事象およびエミシズマブに起因する血栓性マーカーの異常変動は観察されなかった.また,FIXおよびFXの血漿レベルにエミシズマブ投与による影響はみられなかった.エミシズマブの投与下で出血が生じた際には既存の凝固因子製剤が用いられたが,いずれの場合でも良好に止血された.
抗薬物抗体(抗エミシズマブ抗体)は,エミシズマブが投与された健康成人48例中1例で発生した.患者パートにおいては抗薬物抗体による投与中止例はなかったが,他抗体製剤と同様,抗薬物抗体が発生する可能性は否定できないと考えられた.
エミシズマブの国内第I/II相臨床試験(第I相試験患者パートの延長試験)は2013年に,国際共同第III相臨床試験は2015年に開始され,重症血友病A成人患者および小児患者を対象に,週1回,2週に1回または4週に1回のエミシズマブ投与における安全性・忍容性および出血抑制効果等につき,より詳細な検討が進んでいる.本剤は,FVIIIインヒビターの有無に関わらず,週1回の皮下投与により血友病患者における出血を予防できることから,患者やご家族のQOLを著明に向上させると期待される.
また,臨床応用を目指した本研究は,「FVIIIaは,FIXaのプロテアーゼ活性中心をFXの切断部位に正確に配置させることで補因子活性を示す」という仮説を逆に裏づけ,FVIIIの基礎研究に大きな意義をもたらすことができた.
このように,創薬技術の進歩は,今後も,基礎研究を経て臨床に応用され,それが基礎研究に還元され,医療と科学の両方の進歩につながっていくと期待される.
引用文献References
1) Peyvandi, F., Garagiola, I., & Young, G. (2016) Lancet, 388, 187–197.
2) Nilsson, I.M., Berntorp, E., Löfqvist, T., & Pettersson, H. (1992) J. Intern. Med., 232, 25–32.
3) Manco-Johnson, M.J., Abshire, T.C., Shapiro, A.D., Riske, B., Hacker, M.R., Kilcoyne, R., Ingram, J.D., Manco-Johnson, M.L., Funk, S., Jacobson, L., Valentino, L.A., Hoots, W.K., Buchanan, G.R., DiMichele, D., Recht, M., Brown, D., Leissinger, C., Bleak, S., Cohen, A., Mathew, P., Matsunaga, A., Medeiros, D., Nugent, D., Thomas, G.A., Thompson, A.A., McRedmond, K., Soucie, J.M., Austin, H., & Evatt, B.L. (2007) N. Engl. J. Med., 357, 535–544.
4) Lenting, P.J., Donath, M.J., van Mourik, J.A., & Mertens, K. (1994) J. Biol. Chem., 269, 7150–7155.
5) Lapan, K.A. & Fay, P.J. (1997) J. Biol. Chem., 272, 2082–2088.
6) Fay, P.J. & Koshibu, K. (1998) J. Biol. Chem., 273, 19049–19054.
7) Kitazawa, T., Igawa, T., Sampei, Z., Muto, A., Kojima, T., Soeda, T., Yoshihashi, K., Okuyama-Nishida, Y., Saito, H., Tsunoda, H., Suzuki, T., Adachi, H., Miyazaki, T., Ishii, S., Kamata-Sakurai, M., Iida, T., Harada, A., Esaki, K., Funaki, M., Moriyama, C., Tanaka, E., Kikuchi, Y., Wakabayashi, T., Wada, M., Goto, M., Toyoda, T., Ueyama, A., Suzuki, S., Haraya, K., Tachibana, T., Kawabe, Y., Shima, M., Yoshioka, A., & Hattori, K. (2012) Nat. Med., 18, 1570–1574.
8) Sampei, Z., Igawa, T., Soeda, T., Okuyama-Nishida, Y., Moriyama, C., Wakabayashi, T., Tanaka, E., Muto, A., Kojima, T., Kitazawa, T., Yoshihashi, K., Harada, A., Funaki, M., Haraya, K., Tachibana, T., Suzuki, S., Esaki, K., Nabuchi, Y., & Hattori, K. (2013) PLoS One, 8, e57479.
9) Kitazawa, T., Esaki, K., Tachibana, T., Ishii, S., Soeda, T., Muto, A., Kawabe, Y., Igawa, T., Tsunoda, H., Nogami, K., Shima, M., & Hattori, K. (2017) Thromb. Haemost., doi.org/10.1160/TH17-01-0030.
10) Noguchi-Sasaki, M., Soeda, T., Ueyama, A., Muto, A., Shimonaka, Y., Kitamura, H., Kitazawa, T., Fujimoto-Ouchi, K., Kawabe, Y., Nogami, K., Shima, M., & Hattori, K. (2016) Haemophilia, 22(Suppl. 4), 77–78., abstract.
11) Yada, K., Nogami, K., Matsumoto, T., Kitazawa, T., Hattori, K., & Shima, M. (2014) Blood, 124, 4222, abstract.
12) Uchida, N., Sambe, T., Yoneyama, K., Fukazawa, N., Kawanishi, T., Kobayashi, S., & Shima, M. (2016) Blood, 127, 1633–1641.
13) Shima, M., Hanabusa, H., Taki, M., Matsushita, T., Sato, T., Fukutake, K., Fukazawa, N., Yoneyama, K., Yoshida, H., & Nogami, K. (2016) N. Engl. J. Med., 374, 2044–2053.
14) Nogami, K., Soeda, T., Matsumoto, T., Yada, K., Kitazawa, T., Kawabe, Y., Hattori, K., & Shima, M. (2016) Haemophilia, 22(Suppl. 4), 76–77., abstract.
15) Muto, A., Yoshihashi, K., Takeda, M., Kitazawa, T., Soeda, T., Igawa, T., Sakamoto, Y., Haraya, K., Kawabe, Y., Shima, M., Yoshioka, A., & Hattori, K. (2014) J. Thromb. Haemost., 12, 206–213.
16) Muto, A., Yoshihashi, K., Takeda, M., Kitazawa, T., Soeda, T., Igawa, T., Sampei, Z., Kuramochi, T., Sakamoto, A., Haraya, K., Adachi, K., Kawabe, Y., Nogami, K., Shima, M., & Hattori, K. (2014) Blood, 124, 3165–3171.
17) Den Uijl, I.E., Mauser Bunschoten, E.P., Roosendaal, G., Schutgens, R.E., Biesma, D.H., Grobbee, D.E., & Fischer, K. (2011) Haemophilia, 17, 849–853.
著者紹介Author Profile
 北沢 剛久(きたざわ たけひさ)
北沢 剛久(きたざわ たけひさ)中外製薬株式会社研究本部バイオ医薬研究部長.
略歴1993年東京大学農学部獣医学科卒業.同年中外製薬株式会社に入社.以降,抗体医薬の研究に従事.
研究テーマと抱負バイオロジクス医薬品の創製.仲間とともに,今までにない新薬を創製し,患者さんにお届けしたい.
趣味陸上競技(投擲種目).
 嶋 緑倫(しま みどり)
嶋 緑倫(しま みどり)奈良県立医科大学小児科教授.
略歴1979年奈良県立医科大学卒業,同年より奈良県立医科大学小児科研修医,87年米国サンジエゴ市スクリップス研究所リサーチフェロー,89年奈良県立医科大学小児科助手,91年講師,94年助教授,2008年教授,現在に至る.
研究テーマと抱負血液凝固学,血友病の病態.臨床に役に立つ研究.