血管内に形成される血栓は,血管内腔を閉塞し組織を死に至らしめる生死に関わる重大イベントである.血栓形成反応は,血液凝固反応と血小板活性化反応が相互に絡み合っており,これまでに多くの研究が行われ,詳しい機序が明らかになってきている.先進国では,血栓による冠動脈閉塞や脳循環障害が死因の上位に認められ,血栓症の制圧は世界的な喫緊の課題である.また,自然災害が多発する本邦では,災害避難時の深部静脈血栓症および肺塞栓症(静脈血栓が血流にのって肺動脈を閉塞する疾患)の予防が喚起されている.
血栓症とその合併症の発症や再発を予防するため,抗凝固薬や抗血小板薬が広く用いられている.しかし,現在の抗血栓療法は,病的な血栓と生理的な止血の両方に関わる因子(経路)を標的にしているため,出血リスクの増加は避けられない.出血リスクの増加を避けるため,血栓機序と止血機序をあらゆる角度から幅広く解明し,それらの違いを明らかにすることは,止血反応を弱めることなく(すなわち,出血リスクを増加することなく),特異的に動静脈の血栓形成を抑制する新しい戦略を探し出すことにつながると期待されている.
最近,血栓症の研究に生体内イメージング技術やヒト血栓の病因にきわめて類似する動物モデルなどが導入され,これまでに記述されている血液凝固反応と血小板活性化反応に加え,自然免疫系の細胞が血栓の進展と拡大に大きく関わることが次々に報告されてきた1–7).すなわち,これまでの血栓形成機序では大きく取り上げられていなかった白血球が,血栓形成時に活性化され多彩な因子を放出することにより,積極的に病理的な血栓形成に関わることが明らかになり,自然免疫系の細胞が血栓形成に大きく関与するという機序に塗り替えられつつある.また,主に腎臓や脳の細動脈や動脈性毛細血管などの細い血管が血小板血栓により閉塞し,この微小血管の閉塞により腎機能障害や精神神経障害を示す血栓性細小血管症である血栓性血小板減少性紫斑病(thrombotic thrombocytopenic purpura:TTP)の機序も明らかになってきた.
本稿では,古典的な止血機序や動脈血栓の形成機序,およびTTPの発症機序に簡単にふれるとともに,最近明らかになってきた静脈血栓形成機序の新概念を解説し,次いで新しく登場すると考えられるいくつかの抗血栓薬候補の作用機序・標的分子,特に血流環境にフォーカスした新機軸の抗血栓症戦略「ADAMTS13によるフォンビルブランド因子(von Willebrand factor:VWF)機能の制御」の可能性について考察したい.
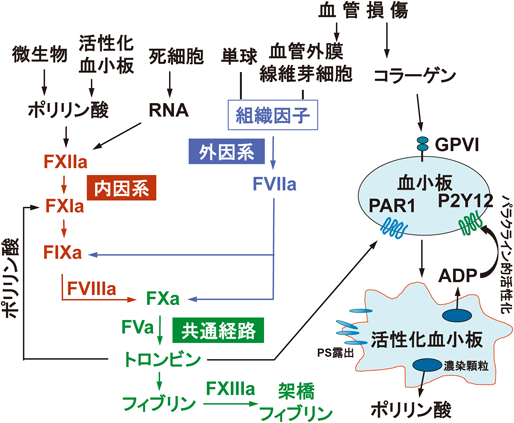
血管が傷ついたときに働く止血機構は,血液の流出を防ぐ生体防衛反応である.止血反応は,血小板が血管損傷部位の内皮下組織に粘着することで始まり,さらに血小板が結合し凝集塊を形成することで血小板血栓(一次止血栓)が完成する.これと平行して,凝固反応が進行しフィブリンが形成され,流血を防ぐ強固な止血栓(二次止血栓)が形成される.血小板凝集は傷口を一時的に閉じるが,凝固は力学的な強度を持つ糊のような物質であるフィブリンの塊で傷口をふさぐ.
1)血小板凝集反応
血小板は血管損傷部位に最初に集まる細胞で,血小板のGPIb-IX-V, GPVI, αIIbβ3が,それぞれ損傷部位のVWF,コラーゲン,フィブリン(フィブリノーゲン)に結合し,多段階で活性化され血小板血栓が形成される.これらの血小板の活性化機序は総説に詳しく述べられている8).血小板は凝固反応で形成されたトロンビンでも活性化される(図1).粘着した血小板は,濃染顆粒から強力な血小板活性化アゴニストであるADPを放出し,パラクライン的な血小板の活性化が起こる9, 10).
2)血液凝固反応
血小板の粘着に平行して,凝固反応が血管傷害部位で活性化される.凝固反応は,膜タンパク質である組織因子で始まり,一連のセリンプロテアーゼとそのコファクターの活性化によりトロンビンが形成されフィブリンの形成につながる(図1)9–11).組織因子は血管壁の外膜の繊維芽細胞や組織の細胞に恒常的に発現している12).外膜の繊維芽細胞は,内膜(血管内皮細胞と基底膜)と中膜(数層の血管平滑筋細胞)により血液との接触が妨げられているため,組織因子による凝固活性化反応は起こらない.血管が損傷を受けると,外膜などにある組織因子が血中のVII(VIIa)に結合できるようになり,凝固反応が起こる.
凝固反応の一つの特徴はポジティブフィードバック活性化,すなわち凝固反応で生成されたトロンビンやXaが上流の因子を活性化することにより,反応が増幅されることにある.初期の凝固反応で生成された微量のトロンビンは,ポジティブフィードバックによりVIIIとVを活性化し,生成したVIIIaとVaはIXaとXaの触媒活性を劇的に亢進する.また,トロンビンはXIも活性化する(図1).XI欠損患者の多くは無症状であり出血症状は比較的軽度であるため,XIは出血リスクを増加させない抗凝固療法の標的因子と考えられている(後述).Xaは組織因子に結合したVIIを活性化する.トロンビンはフィブリノーゲンをフィブリンに変換し,トランスグルタミナーゼであるXIIIを活性化し,最終的に,XIIIaにより安定化したフィブリンからなる力学的に強固な止血栓を形成する.
3)血小板凝集と凝固反応の相互作用
凝固反応はトロンビンを生成し,トロンビンはprotease-activated receptor(PAR,ヒトではPAR1,マウスではPAR4)を介して血小板を活性化させる(図1).トロンビンやコラーゲンにより活性化した血小板は,プロトロンビン,VII, IX, Xが親和性を有する酸性リン脂質であるホスファチジルセリンを細胞膜上に露出し,凝固反応の場を提供することにより反応を促進する(図1).活性化した血小板は,内部の濃染顆粒からポリリン酸を放出する.ポリリン酸はトロンビンによるXI活性化の促進作用などを持ち(後述),凝固反応を増幅する.このように,血小板の活性化と凝固反応は互いに支え合いながら機能を増強している.
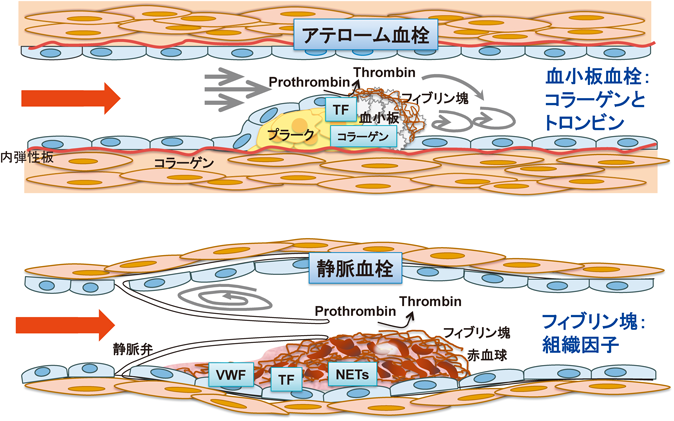
動脈血栓症と静脈血栓症は発生や機序が異なる疾患である(図2).動脈血栓症は,主に粥状プラークの破裂によって起こるのでアテローム血栓症と呼ばれる13).アテローム血栓症では,内皮下層に形成された脂質に富んだプラークに,組織因子を発現する単球や平滑筋細胞,また壊死を起こした細胞が局在する.プラークは薄い単層の血管内皮細胞で血液と隔てられている.この繊維状キャップが破綻やびらんを起こしはがれ,下層の細胞外マトリクスのコラーゲンが血流にさらされると,血小板の活性化が進行し血小板粘着が起こるとともに,組織因子により凝固反応が開始しフィブリン塊が形成される.
コラーゲンで活性化された血小板は,濃染顆粒からADPなどの血小板活性化物質を放出し,トロンボキサンA2を速やかに合成して放出する.トロンビンも血小板を活性化する.これらのアゴニストは,特異的な受容体を介して血小板を活性化し,血小板インテグリンαIIbβ3の活性化につながる9, 10).粘着した血小板は血流に乗って流れてくる血小板を次々と捕獲・結合し(血小板凝集反応),血小板血栓が空間的に成長する.
静脈血栓症では血管内皮細胞は活性化している(図3)1, 2, 4, 14).最近,マウスの下大静脈狭窄モデルが開発され,血流が低下した条件下の静脈での血栓形成機序が詳しく解析された1).このマウスモデルによると,内腔を80%程度狭窄した48時間後の静脈内に血栓が観察されている.これは血流の低下により内皮細胞が活性化され,接着因子であるVWFが係留し,かつ内皮表面にPセレクチンを発現し,これらの接着因子に単球や好中球が結合し活性化され,積極的に凝固反応が開始され拡大し,また血小板が活性化することにより,さらに凝固反応が増幅されることを示している.静脈血栓は単球や好中球といった自然免疫系に関わる細胞が関与するので,2013年EngelmannとMassbergはこれをImmunothrombosisと呼び,これらの細胞が活性化される際に放出される無菌性炎症(sterile inflammation)に関わる因子群が,静脈血栓を増大させることを示した5).また,最近では虚血性の脳血管傷害もImmunothrombosisが大きく関わることが明らかになっている15).
静脈血栓の形成は多段階からなる(図3).静脈血栓形成は多くの反応が同時進行すると考えられるため,各段階を明確に区分することはできないが,便宜上,いくつかの段階に分けてそれぞれの特徴を示したい.
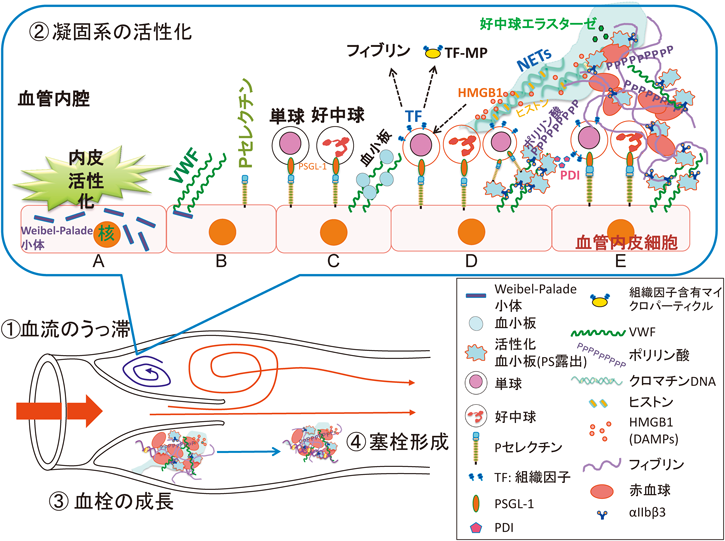
1)ステージA:
静脈血栓が生じる血管内の局所では,血管内皮細胞は形態学的には無傷のままであり傷害はみられないが,血液のうっ滞により内皮細胞は活性化され,炎症性の表現型を獲得していると考えられている16).静脈血栓は静脈弁で起こると考えられている16).剖検と静脈造影の所見で,静脈弁のポケットで血栓が形成されることが示されている.静脈弁は下肢の血液の循環を補助する働きがあり,起立時,1分間に20回程度開閉を行っている17).弁の裏(弁ポケット)では血流は渦状に流れ,弁ポケット内はうっ血で低酸素状態になりやすい場所である(図3)16, 18).弁のポケット側の内皮細胞は外側の内皮細胞に比べ,抗凝固タンパク質であるEPCRとトロンボモジュリンが多く発現し,VWFは少ないという免疫染色の結果が報告されており,弁ポケット内を血栓抵抗性に維持する工夫がみられる16, 19).
静脈血栓症のリスク因子として長期臥床や不動が知られている20).これは静止状態により静脈弁が働かないことにより血液のうっ滞が起こりやすくなると考えられている.同じようにうっ血が起こりやすい部位として,心臓左心房の袋状構造をとる心耳があげられる.心耳に血栓が生じると,血流にのって脳血管へと流れ,心原性脳塞栓症の原因になる.したがって,心原性脳塞栓症の再発予防には,抗凝固薬が用いられる.
2)ステージB:
活性化された内皮細胞は,細胞内のWeibel-Palade小体に蓄えられている膜タンパク質であるPセレクチンを細胞膜上に移動させ,VWFを循環血中に放出するとともに,一部のVWFを細胞膜上に係留する.Weibel-Palade小体には,2050アミノ酸残基からなるVWFモノマーがC末端領域間,およびN末端領域間でジスルフィド結合し,200以上のモノマーがつながった超高分子量VWFマルチマーとして貯蔵されている.この巨大分子はきわめて血小板凝集能が高い.これが血中に分泌されるとVWF切断酵素ADAMTS13で限定分解を受け,40から200程度のモノマーからなる適当な血小板凝集能を持つマルチマーとなる21)(後述,秋山の稿を参照).マウスの下大静脈の狭窄モデルでは,狭窄6時間後に血管内皮細胞のWeibel-Palade小体が消失(VWFの放出とPセレクチンの細胞上への移動)し,内皮上に血小板と白血球の集積が観察されている2).
3)ステージC:
活性化内皮細胞表面の接着因子に,循環血中の好中球や単球,血小板が結合する1).活性化内皮細胞や活性化血小板の表面のPセレクチンには,P-selectin glycoprotein ligand 1(PSGL-1)を発現する好中球や単球が結合する.好中球が早く集積し,続いて単球の集積が観察され,48時間後の静脈血栓中の白血球の約70%は好中球で約30%が単球であるという1).一方,内皮細胞上に係留するVWFには,膜タンパク質であるGPIbを介して血小板が結合する.
4)ステージD:
内皮細胞に集積した単球,好中球,血小板は,固有の活性化経路により異なった反応を行いつつ血栓形成に寄与する22).単球はdamage-associated molecular patterns(DAMPs,損傷関連分子パターン)により刺激依存性に組織因子を発現し,一連のプロテアーゼとタンパク質性補助因子の活性化を通してトロンビンを産生しフィブリン塊を形成する.
VWFに結合した血小板はトロンビンにより活性化され,ADPやトロンボキサンA2,ポリリン酸の放出,ホスファチジルセリンの露出などすでに述べたような機序を通して血栓形成に大きく関わるとともに,Pセレクチンを介して白血球の集積にも寄与する1).血小板由来のポリリン酸は,60~100個のリン酸が直鎖につながった陰性荷電を持つ物質で,トロンビンによるXIのフィードバック活性化の促進,XaとトロンビンによるVの活性化の促進,組織因子経路インヒビター(TFPI)活性の減弱といった血栓促進作用を有し,血栓の拡大につながる働きをする23).このポリリン酸の内因系凝固の活性化能は低いと考えられている.
集積した好中球は,核内のクロマチンDNA,ヒストン,high-mobility group box 1(HMGB1),および顆粒内のプロテアーゼや殺菌物質などからなるneutrophil extracellular traps(NETs)を放出し,XIIを結合し血小板と赤血球を絡めつつ血栓形成を促進する1, 6).NETsおよびそれとともに放出されるRNAは,ともに陰電荷の性質を利用して内因系凝固因子であるXIIとXIを活性化させる24).NETsは感染と炎症の場に凝固反応を局在化させる14).顆粒から放出された好中球エラスターゼは,抗凝固活性を持つTFPIを分解・失活させ凝固を促進する14).放出されたヒストンは血小板凝集を惹起し25),血小板を刺激してトロンビン産生能を亢進し26),赤血球に働きホスファチジルセリンを表面に露出させ,凝固反応を促進する作用を示す27).
集積した単球,好中球,血小板が活性化して放出する因子は複雑に作用しつつ静脈血栓を拡大する働きを持つ.最近,代表的なDAMPsとして知られるHMGB1は,血栓形成にきわめて重要な働きを示すことが明らかになった28, 29).血小板特異的HMGB1欠損マウスは出血時間の延長,血栓量の減少,血小板凝集能の低下,炎症の低下,実験的外傷での組織損傷の低下が観察された28).また,静脈血栓形成の際に血小板から放出されるHMGB1は還元型HMGB1であり主に単球を集積する役割を果たし,活性化好中球により酸化型HMGB1となり,これが血小板凝集を行うことでさらにHMGB1量を増加させる.また,酸化型HMGB1は単球を活性化し組織因子含有マイクロパーティクルと炎症性サイトカインを産生し,NETs形成を行うと報告された29).好中球が放出するNETsに関しては,血中を循環した好中球(aged neutrophil)は,骨髄から新たに放出された好中球より,NETsを放出する能力が高いと報告された30, 31).
protein disulfide isomerase(PDI)を阻害すると,血栓形成が抑制されることから,血栓形成に関わる分子の酸化還元状態が血栓形成に関与すると考えられている32, 33).PDIは,活性化血小板,血管内皮細胞,障害を受けた細胞から分泌され,血栓形成に働くと考えられているが,その機序は不明な点が多い.血管内の組織因子のほとんどは,機能的に不活性型であり,PDIで活性化される必要があるとの報告がある34).組織因子の膜に近いジスルフィド結合(C186-C209間)は酸化還元を受けやすい.インテグリンαIIbβ3もPDIの標的酵素であるとの報告がある32).
5)ステージE:
フィブリンとVWFはRGD配列を介して活性型の立体構造をとるインテグリンαIIbβ3に結合し血小板凝集を引き起こす.フィブリンはXIIIaの作用によりγ鎖間で架橋されたγダイマーとα鎖間で架橋されたα鎖ポリマーとなり,力学的に強固なフィブリンポリマーへと変換される.赤血球はフィブリンα鎖ポリマーにトラップされて静脈血栓中に存在する35).これらの反応の結果,静脈血栓には血小板や赤血球,白血球が混在する.血栓形成の足場としてフィブリン,VWFマルチマー,NETsが存在する.このように考えると,フィブリンを溶かすプラスミン,VWF切断酵素であるADAMTS13, NETsの主体であるDNAを分解するDNA分解酵素が,血栓の足場を溶かす酵素といえよう.
TTP病態の基盤である微小血管における血小板凝集は,VWFが決定的な役割を果たしている.TTPはVWF切断酵素であるADAMTS13の著しい活性低下(臨床的には正常値の10%未満)で惹起される36, 37).TTPの舞台である微小循環系は,大血管とは異なり流体力学的な力である“ずり応力”がきわめて高く,特徴的な血流動態を示すことが知られている.
1)ずり応力とVWF機能
血管内では一般的に,赤血球は血管内腔の中心部を比較的早いスピードで流れ,血小板や白血球などは血管壁近傍を比較的遅いスピードで流れている.このスピードの差異が流体力学的なストレス(ずり応力,shear stress)を生じさせる.ずり応力は血管径に反比例し血流速度に比例するため,血液がゆっくり流れている大きな静脈などでは,血小板などの血球成分はたいしたストレスを受けない.逆に,血液が速く流れている細動脈や血管径の細い動脈性毛細血管などではきわめて高いずり応力が発生していると想定される.
VWFは高ずり速度下で血小板の粘着・凝集機能をいかんなく発揮する38, 39).このVWFの高ずり応力依存性の機能特性は,そのユニークなマルチマー構造によるものと考えられている.前述のように,VWFはモノマーが直鎖上につながったマルチマー構造をとり,マルチマー重合度の高いVWFほど血小板粘着・凝集活性が高い.静止下ではVWFマルチマーは球状構造をとりVWFの血小板結合部位,特にGPIb結合部位は球状構造の内部に潜在しているが,ずり応力がかかるとこの部位が露出すると想定され,ずり応力が高くなればなるほど血小板粘着・凝集活性が増強する「VWFのずり応力依存性機能」の特性を説明するものと考えられている.
2)高ずり応力下でのVWF-ADAMTS13軸の機能連関
先に述べたように,高ずり応力下でのVWFの血小板粘着・凝集機能はマルチマー構造に依存するため,マルチマー重合度の低いVWFは血小板粘着・凝集機能が弱く,結果として出血傾向を示す.逆に,マルチマー重合度が高すぎる超高分子量VWFマルチマーの場合,TTPを引き起こす40).したがって,VWFマルチマーは生体では適度なサイズに制御されていると考えられ,この働きを担っているのがADAMTS13である.すなわち,ADAMTS13活性が著しく低下すると超高分子量VWFマルチマーが血中に循環し,高いずり応力を示す微小血管内にVWFを足場にした血小板血栓が生じる.これがTTPの発症機序と考えられる.
生体内でのADAMTS13によるVWFの切断機序は,ながらく不明であった.最近我々はADAMTS13の機能もVWFマルチマーと同様,ずり応力依存性であることを明らかにした41).すなわち,高ずり応力でVWFマルチマーが引き伸ばされることにより,切断部位であるVWF A2ドメインがより露出して変性し,VWF切断活性が増強すると推定された.
VWFは血管内皮細胞から放出された直後は,血小板凝集能が非常に強い超高分子量VWFマルチマーであるが,ADAMTS13によって速やかに適正なマルチマーサイズに切断され,血液中を循環し始めると考えられている.また,これに加えて,ADAMTS13は高ずり応力下での血栓形成現場で血栓形成に参画しているVWFを切断し,リアルタイムにVWF機能ならびに血栓成長を制御していることも判明した41).
3)VWF-ADAMTS13軸のずり応力依存性機能特性に基づく血栓形成の制御
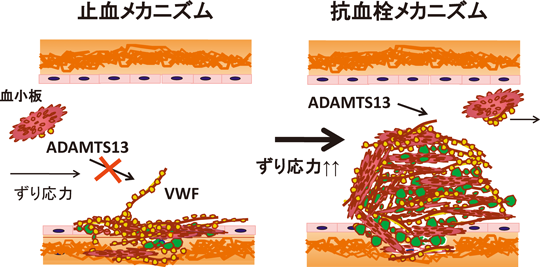
ずり応力で,まずVWF-GPIb相互作用が促進され,次いでVWF依存性の血小板粘着・凝集が進行する.これは止血の際に観察される(止血メカニズム).血小板粘着・凝集の初期相のこの時点ではADAMTS13の機能はいまださほど活発ではない(図4).血小板粘着・凝集反応の進行につれ,徐々に血栓が大きくなっていき,血管内腔のフリースペースが狭小化することになる.前述したずり応力の定義「血管内腔が狭いほどずり応力が高い」から考えると,結果として血栓の増大に呼応して局所ずり応力が高くなる.理論的には,閉塞直前の血管内腔におけるずり応力は無限大に上昇しているはずである.同時に,血流に直接曝露する血栓成長先進部では,VWFへの活発な血小板結合でADAMTS13によるVWF切断活性が飛躍的に亢進する(図4).いい換えれば,血栓が成長すればするほど,ADAMTS13の血栓成長ストッパー(制御)機能が増幅されて致命的な血管閉塞を回避させることになる(抗血栓メカニズム).
このように,ADAMTS13がずり応力に依存してVWFマルチマーを分解し,その結果血小板血栓形成が抑制されて血管の閉塞に抵抗する特性は,止血には影響を与えないきわめてユニークな性質であり,このことからADAMTS13はずり応力がかかる動脈での血栓を抑制する可能性を示唆している.
脳梗塞,心筋梗塞,末梢動脈疾患はアテローム血栓症であり血小板血栓なので,再発予防には抗血小板薬であるアスピリンやチエノピリジン系抗血小板薬が用いられる.アスピリンは,血小板のシクロオキシゲナーゼ1を不可逆的に阻害し,トロンボキサンA2の産生を抑制することにより抗血小板作用を発揮する.チエノピリジン系抗血小板薬は,血小板アゴニストであるADPの特異的受容体であるP2Y12に共有結合し,そのシグナル伝達を阻止することにより,血小板の活性化を抑制する.TTPはADAMTS13の活性著減により発症するため,血漿交換でADAMTS13の補充と活性を中和する抗体の除去を行う.
静脈血栓症や肺塞栓症,心原性脳塞栓症は凝固系が主体のフィブリン血栓であり,治療は抗凝固療法で行われる.抗凝固薬として,注射薬のヘパリン類,経口薬で古くから使われているワルファリンと,最近使用され始めたdirect oral anticoagulants(DOACs)がある.DOACには抗トロンビン薬と抗Xa薬がある.これらの現在使用されている抗血栓薬は,いずれも病的な血栓と生理的な止血の両方に関わる因子(経路)を標的にしているため,出血リスクを伴う.ここでは,これまで述べてきた血栓機序に基づくいくつかの新規抗血栓薬候補の研究成果について述べる.
1)XI, XII
注目されている抗凝固薬の標的因子は内因系凝固に関わるXIとXIIである42).前述したように,XI欠損患者の多くは無症状で出血症状は比較的軽度であるため,薬剤で阻害しても重大な出血事象は起こりにくいと考えられる43).XIはトロンビンがフィードバック活性化を行うので血栓形成に寄与すると考えられ,ノックアウトマウスを使った研究で,XIは病的血栓形成に重要な役割を果たしていることが明らかになってきた43).外科手術では,組織が破壊されて組織因子が露出し,破壊された細胞や活性化血小板などからポリリン酸やRNA, DAMPsが放出され,凝固系が作動すると考えられる.XIを標的としたアンチセンスオリゴヌクレオチドは,肝細胞内のXI mRNAを分解し,循環血中のXI量を減少させ,内因系凝固反応を抑制する.2015年,膝関節置換術患者にXIのアンチセンスオリゴヌクレオチドを術前に皮下注射し,術後の静脈血栓症発症を調べた結果が報告された44).XI活性を約38%低下させた患者群では,標準抗凝固療法の患者群と比較し静脈血栓発症は非劣性だったが,約20%にまで低下させた患者群では優れた血栓抑制能を示した.この研究は,XI量を低下させると術後の血栓塞栓症例が減るという結果を示しており,大変注目に値する.XIもしくはXIIを標的にした薬剤がより理想的な抗血栓薬になる可能性が議論されている42).
2)VWFとADAMTS13
血小板とVWFの結合を阻害するVWFのA1ドメインを標的にする抗体薬が開発され,ADAMTS13自己抗体で発症する後天性TTPを対象にした成果が発表された45).
a.脳梗塞におけるVWF-ADAMTS13軸
血栓性細小血管症であるTTPにおけるVWF-ADAMTS13軸の重要な貢献は明らかとなったが36, 37),脳梗塞や心筋梗塞などの動脈血栓症においてはどうであろうか?
ここ数年来,マウスモデルの研究で,VWF-ADAMTS13軸が脳梗塞の病態形成・進展に大きく関与することが明らかになった.すなわち,実験的脳虚血/再灌流モデルにおいて,①VWF-ノックアウト(KO)マウスは,野生型に比して梗塞体積が有意に縮小している,②逆に,超高分子量VWFマルチマーを持つADAMTS13-KOマウスでは脳梗塞は有意に悪化する,③ADAMTS13-KOマウスにおける脳梗塞の増悪はADAMTS13の投与で野生型レベルにまで改善する,という成績が我々のグループをはじめ次々と報告され,ADAMTS13の脳神経保護作用が注目されるようになった46–48).さらに注目すべきは,これら脳虚血再灌流障害モデルに加えて,くも膜下出血モデルによる脳虚血病態が,ADAMTS13の投与で著明に改善することが報告された49).これは,ADAMTS13によるVWF依存性微小血栓形成の抑制が,脳微小循環の改善をもたらすためと考えられた.
いわゆる従来型の抗血栓薬は出血を助長するので,くも膜下出血や出血性梗塞のような出血を基盤とした脳虚血性疾患には使えない.これに対して,前述のようにADAMTS13の脳神経保護作用は理論的には出血を助長しないので,広く脳虚血・梗塞性疾患の治療選択肢としてきわめて有望と考えられる.
しかしながら,これらのマウス脳梗塞モデルでの実験成績は,「VWF-ADAMTS13軸が脳梗塞・脳動脈血栓症の成立メカニズムに直結する」との結論を必ずしも支持するわけではない.あくまで,実験的にマウスに脳虚血による脳梗塞を誘起した場合,VWF-ADAMTS13軸が病態形成・進展に大きく関与することが判明したのである.
b.心筋梗塞におけるVWF-ADAMTS13軸
脳梗塞と並んで二大動脈血栓症ともいえる心筋梗塞についても,VWF-ADAMTS13軸の重要性が我々や他のグループから報告された50–52).結論から述べると,脳梗塞の場合とほぼ同様の成績,つまり心筋保護作用がマウス心筋梗塞モデル実験でも確認された.
De Meyerらは,心筋虚血/再灌流(0.5時間虚血+23.5時間再灌流)の実験系で,ADAMTS13-KOマウスでは野生型に比して有意に梗塞体積が増大することを見いだした50).また,野生型マウスに組換えヒトADAMTS13(rhADAMTS13)を前処置投与した場合,梗塞体積が有意に減少することも報告している.また,Gandhiらは,完全に同一の虚血/再灌流実験で,rhADAMTS13投与成績は示さなかったが,ADAMTS13-KOマウスにおける同様の成績に加えてVWF-KOマウスの成績を報告している51).すなわち,VWF-KOマウスではADAMTS13-KOとは逆に,心筋梗塞体積が縮小していることを示した.ここまでに報告された脳梗塞モデルの実験や,今回のDe Meyerら,Gandhiらの心筋梗塞の成績はいずれもすべて虚血/再灌流の実験条件下でなされている50, 51).虚血/再灌流障害の発生メカニズムに関してはいまだ不明な点が多いが,再灌流時の過剰炎症機転が重要な役割を果たすと考えられており,あらためてADAMTS13の抗炎症作用の重要性が明らかとなった.
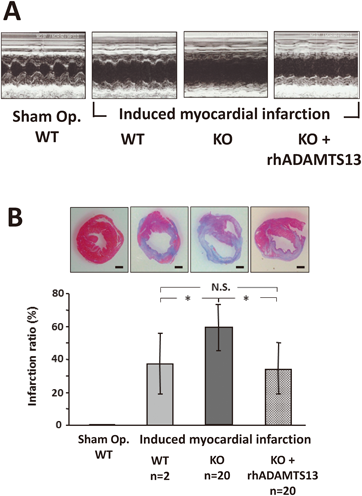
これらに対して,我々は虚血のみで再還流を誘導しない心筋梗塞モデルを用いた52).マウス冠動脈左前下行枝を結紮して心筋梗塞を誘起し,1週後のマウス心機能をMモード心エコーで評価したのち,心筋梗塞を組織学的に検討した.その結果,野生型と比較してADAMTS13-KOマウスで有意な心機能低下と梗塞範囲の増大を認めた(図5).これらADAMTS13-KOマウスの心所見は,ADAMTS13の投与で野生型レベルにまで改善された(図5).この結果より,ADAMTS13の虚血時の心筋保護作用は必ずしも虚血/再灌流障害に限定されないことが明らかとなった.さらに特記すべきは,我々の実験結果はADAMTS13を前投与するのではなく,心筋梗塞誘起後の投与で心筋保護効果が観察された点であり,治療応用ポテンシャルの高さを示唆している.しかしながら,ブタの心筋梗塞モデルに遺伝子組換えヒトADAMTS13を投与しても改善効果を認めなかったとの報告もあり53),ADAMTS13の心筋梗塞における心筋保護機能の作用メカニズムに関してはいまだ解明すべき点が多い.
c.静脈血栓におけるVWF-ADAMTS13軸
ADAMTS13-KOマウスは,電気分解による下大静脈傷害性の静脈血栓モデルにおいて,野生型マウスに比べて大きな静脈血栓が生じ,ADAMTS13投与により血栓量の減少を認めた54).ADAMTS13の遺伝子欠損により,超高分子量VWFマルチマーが静脈血栓部位により多くの血小板,単球,好中球をリクルートするためと考えられる.
凝固反応と血小板活性化という古典的な血栓形成機序に加え,最近明らかになってきた白血球の関与を取り入れた新しい血栓形成機序を概説し,それに沿った我々の研究を紹介した.血栓は時間とともにダイナミックにまた空間的に変化する魅力的な研究対象であり,その成果がヒト疾患の解明と新規の薬剤標的につながる可能性を持っている.
引用文献References
1) von Bruhl, M.L., Stark, K., Steinhart, A., Chandraratne, S., Konrad, I., Lorenz, M., Khandoga, A., Tirniceriu, A., Coletti, R., Kollnberger, M., Byrne, R.A., Laitinen, I., Walch, A., Brill, A., Pfeiler, S., Manukyan, D., Braun, S., Lange, P., Riegger, J., Ware, J., Eckart, A., Haidari, S., Rudelius, M., Schulz, C., Echtler, K., Brinkmann, V., Schwaiger, M., Preissner, K.T., Wagner, D.D., Mackman, N., Engelmann, B., & Massberg, S. (2012) J. Exp. Med., 209, 819–835.
2) Brill, A., Fuchs, T.A., Chauhan, A.K., Yang, J.J., De Meyer, S.F., Kollnberger, M., Wakefield, T.W., Lammle, B., Massberg, S., & Wagner, D.D. (2011) Blood, 117, 1400–1407.
3) Brill, A., Fuchs, T.A., Savchenko, A.S., Thomas, G.M., Martinod, K., De Meyer, S.F., Bhandari, A.A., & Wagner, D.D. (2012) J. Thromb. Haemost., 10, 136–144.
4) Mackman, N. (2012) J. Clin. Invest., 122, 2331–2336.
5) Engelmann, B. & Massberg, S. (2013) Nat. Rev. Immunol., 13, 34–45.
6) Martinod, K. & Wagner, D.D. (2014) Blood, 123, 2768–2776.
7) Gaertner, F. & Massberg, S. (2016) Semin. Immunol., 28, 561–569.
8) Denis, C.V. & Wagner, D.D. (2007) Arterioscler. Thromb. Vasc. Biol., 27, 728–739.
9) Furie, B. & Furie, B.C. (2008) N. Engl. J. Med., 359, 938–949.
10) Versteeg, H.H., Heemskerk, J.W., Levi, M., & Reitsma, P.H. (2013) Physiol. Rev., 93, 327–358.
11) Wolberg, A.S., Aleman, M.M., Leiderman, K., & Machlus, K.R. (2012) Anesth. Analg., 114, 275–285.
12) Drake, T.A., Morrissey, J.H., & Edgington, T.S. (1989) Am. J. Pathol., 134, 1087–1097.
13) Libby, P. (2013) N. Engl. J. Med., 368, 2004–2013.
14) Massberg, S., Grahl, L., von Bruehl, M.L., Manukyan, D., Pfeiler, S., Goosmann, C., Brinkmann, V., Lorenz, M., Bidzhekov, K., Khandagale, A.B., Konrad, I., Kennerknecht, E., Reges, K., Holdenrieder, S., Braun, S., Reinhardt, C., Spannagl, M., Preissner, K.T., & Engelmann, B. (2010) Nat. Med., 16, 887–896.
15) De Meyer, S.F., Denorme, F., Langhauser, F., Geuss, E., Fluri, F., & Kleinschnitz, C. (2016) Stroke, 47, 1165–1172.
16) Bovill, E.G. & van der Vliet, A. (2011) Annu. Rev. Physiol., 73, 527–545.
17) Aird, W.C. (2007) J. Thromb. Haemost., 5(Suppl 1), 283–291.
18) Esmon, C.T. (2009) Blood Rev., 23, 225–229.
19) Brooks, E.G., Trotman, W., Wadsworth, M.P., Taatjes, D.J., Evans, M.F., Ittleman, F.P., Callas, P.W., Esmon, C.T., & Bovill, E.G. (2009) Blood, 114, 1276–1279.
20) Tapson, V.F. (2008) N. Engl. J. Med., 358, 1037–1052.
21) Springer, T.A. (2014) Blood, 124, 1412–1425.
22) Swystun, L.L. & Liaw, P.C. (2016) Blood, 128, 753–762.
23) Morrissey, J.H. (2012) Int. J. Hematol., 95, 346–352.
24) Zernecke, A. & Preissner, K.T. (2016) Circ. Res., 118, 469–479.
25) Fuchs, T.A., Bhandari, A.A., & Wagner, D.D. (2011) Blood, 118, 3708–3714.
26) Semeraro, F., Ammollo, C.T., Morrissey, J.H., Dale, G.L., Friese, P., Esmon, N.L., & Esmon, C.T. (2011) Blood, 118, 1952–1961.
27) Semeraro, F., Ammollo, C.T., Esmon, N.L., & Esmon, C.T. (2014) J. Thromb. Haemost., 12, 1697–1702.
28) Vogel, S., Bodenstein, R., Chen, Q., Feil, S., Feil, R., Rheinlaender, J., Schaffer, T.E., Bohn, E., Frick, J.S., Borst, O., Munzer, P., Walker, B., Markel, J., Csanyi, G., Pagano, P.J., Loughran, P., Jessup, M.E., Watkins, S.C., Bullock, G.C., Sperry, J.L., Zuckerbraun, B.S., Billiar, T.R., Lotze, M.T., Gawaz, M., & Neal, M.D. (2015) J. Clin. Invest., 125, 4638–4654.
29) Stark, K., Philippi, V., Stockhausen, S., Busse, J., Antonelli, A., Miller, M., Schubert, I., Hoseinpour, P., Chandraratne, S., von Bruhl, M.L., Gaertner, F., Lorenz, M., Agresti, A., Coletti, R., Antoine, D.J., Heermann, R., Jung, K., Reese, S., Laitinen, I., Schwaiger, M., Walch, A., Sperandio, M., Nawroth, P.P., Reinhardt, C., Jackel, S., Bianchi, M.E., & Massberg, S. (2016) Blood, 128, 2435–2449.
30) Zhang, D., Chen, G., Manwani, D., Mortha, A., Xu, C., Faith, J.J., Burk, R.D., Kunisaki, Y., Jang, J.E., Scheiermann, C., Merad, M., & Frenette, P.S. (2015) Nature, 525, 528–532.
31) Uhl, B., Vadlau, Y., Zuchtriegel, G., Nekolla, K., Sharaf, K., Gaertner, F., Massberg, S., Krombach, F., & Reichel, C.A. (2016) Blood, 128, 2327–2337.
32) Furie, B. & Flaumenhaft, R. (2014) Circ. Res., 114, 1162–1173.
33) Schulman, S., Bendapudi, P., Sharda, A., Chen, V., Bellido-Martin, L., Jasuja, R., Furie, B.C., Flaumenhaft, R., & Furie, B. (2016) Antioxid. Redox Signal., 24, 1–15.
34) Reinhardt, C., von Bruhl, M.L., Manukyan, D., Grahl, L., Lorenz, M., Altmann, B., Dlugai, S., Hess, S., Konrad, I., Orschiedt, L., Mackman, N., Ruddock, L., Massberg, S., & Engelmann, B. (2008) J. Clin. Invest., 118, 1110–1122.
35) Byrnes, J.R., Duval, C., Wang, Y., Hansen, C.E., Ahn, B., Mooberry, M.J., Clark, M.A., Johnsen, J.M., Lord, S.T., Lam, W.A., Meijers, J.C., Ni, H., Ariens, R.A., & Wolberg, A.S. (2015) Blood, 126, 1940–1948.
36) Furlan, M., Robles, R., Galbusera, M., Remuzzi, G., Kyrle, P.A., Brenner, B., Krause, M., Scharrer, I., Aumann, V., Mittler, U., Solenthaler, M., & Lammle, B. (1998) N. Engl. J. Med., 339, 1578–1584.
37) Tsai, H.M. & Lian, E.C. (1998) N. Engl. J. Med., 339, 1585–1594.
38) Tsuji, S., Sugimoto, M., Miyata, S., Kuwahara, M., Kinoshita, S., & Yoshioka, A. (1999) Blood, 94, 968–975.
39) Ruggeri, Z.M. (2000) J. Clin. Invest., 105, 699–701.
40) Sadler, J.E. (2005) J. Thromb. Haemost., 3, 1702–1709.
41) Shida, Y., Nishio, K., Sugimoto, M., Mizuno, T., Hamada, M., Kato, S., Matsumoto, M., Okuchi, K., Fujimura, Y., & Yoshioka, A. (2008) Blood, 111, 1295–1298.
42) Fredenburgh, J.C., Gross, P.L., & Weitz, J.I. (2017) Blood, 129, 147–154.
43) Lowenberg, E.C., Meijers, J.C., Monia, B.P., & Levi, M. (2010) J. Thromb. Haemost., 8, 2349–2357.
44) Buller, H.R., Bethune, C., Bhanot, S., Gailani, D., Monia, B.P., Raskob, G.E., Segers, A., Verhamme, P., & Weitz, J.I., FXI-ASO TKA Investigators. (2015) N. Engl. J. Med., 372, 232–240.
45) Peyvandi, F., Scully, M., Kremer Hovinga, J.A., Cataland, S., Knobl, P., Wu, H., Artoni, A., Westwood, J.P., Mansouri Taleghani, M., Jilma, B., Callewaert, F., Ulrichts, H., Duby, C., Tersago, D., & Investigators, T., TITAN Investigators. (2016) N. Engl. J. Med., 374, 511–522.
46) Zhao, B.Q., Chauhan, A.K., Canault, M., Patten, I.S., Yang, J.J., Dockal, M., Scheiflinger, F., & Wagner, D.D. (2009) Blood, 114, 3329–3334.
47) Kleinschnitz, C., De Meyer, S.F., Schwarz, T., Austinat, M., Vanhoorelbeke, K., Nieswandt, B., Deckmyn, H., & Stoll, G. (2009) Blood, 113, 3600–3603.
48) Fujioka, M., Hayakawa, K., Mishima, K., Kunizawa, A., Irie, K., Higuchi, S., Nakano, T., Muroi, C., Fukushima, H., Sugimoto, M., Banno, F., Kokame, K., Miyata, T., Fujiwara, M., Okuchi, K., & Nishio, K. (2010) Blood, 115, 1650–1653.
49) Muroi, C., Fujioka, M., Mishima, K., Irie, K., Fujimura, Y., Nakano, T., Fandino, J., Keller, E., Iwasaki, K., & Fujiwara, M. (2014) J. Thromb. Haemost., 12, 505–514.
50) De Meyer, S.F., Savchenko, A.S., Haas, M.S., Schatzberg, D., Carroll, M.C., Schiviz, A., Dietrich, B., Rottensteiner, H., Scheiflinger, F., & Wagner, D.D. (2012) Blood, 120, 5217–5223.
51) Gandhi, C., Motto, D.G., Jensen, M., Lentz, S.R., & Chauhan, A.K. (2012) Blood, 120, 5224–5230.
52) Doi, M., Matsui, H., Takeda, H., Saito, Y., Takeda, M., Matsunari, Y., Nishio, K., Shima, M., Banno, F., Akiyama, M., Kokame, K., Miyata, T., & Sugimoto, M. (2012) Thromb. Haemost., 108, 1236–1238.
53) Eerenberg, E.S., Teunissen, P.F., van den Born, B.J., Meijers, J.C., Hollander, M.R., Jansen, M., Tijssen, R., Belien, J.A., van de Ven, P.M., Aly, M.F., Kamp, O., Niessen, H.W., Kamphuisen, P.W., Levi, M., & van Royen, N. (2016) Cardiovasc. Res., 111, 194–203.
54) Tashima, Y., Banno, F., Akiyama, M., & Miyata, T. (2015) Thromb. Haemost., 114, 206–207.
著者紹介Author Profile
 宮田 敏行(みやた としゆき)
宮田 敏行(みやた としゆき)国立研究開発法人国立循環器病研究センター脳血管内科シニア研究員.理学博士.
略歴1954年京都府に生る.83年九州大学大学院理学研究科博士終了.83~86年日本学術振興会研究員.86年九州大学理学部助手.91年国立循環器病(研究)センター室長(2001年~部長,13年~部門長).15年~現職.
研究テーマと抱負血栓症,特に静脈血栓症,血栓性細小血管症の基礎研究を臨床に繋げたい.
 樋口(江浦) 由佳(えうら ゆか)
樋口(江浦) 由佳(えうら ゆか)国立研究開発法人国立循環器病研究センター分子病態部上級研究員.博士(理学).
略歴兵庫県出身.2001年九州大学理学部卒.06年同大学システム生命科学府博士課程修了.06年~国立循環器病センター病因部・流動研究員.08年~日本学術振興会・特別研究員PD.11年~国立循環器病研究センター分子病態部・流動研究員.その後非常勤研究員等を経て16年より現職.
研究テーマと抱負VWFマルチマーに関する研究とTTP患者の遺伝子解析,小胞体関連分解(ER-associated degradation)に関する研究.初心を忘れず情熱をもって研究者道を邁進したい.
ウェブサイトhttp://www.ncvc.go.jp/res/divisions/etiology/index.html
趣味美味しいものの食べ歩き.自然豊かな寺社仏閣や公園の散策.娘の写真を撮ること.ラテン音楽鑑賞とラテンダンス.
 杉本 充彦(すぎもと みつひこ)
杉本 充彦(すぎもと みつひこ)奈良県立医科大学血栓制御医学教授.医学博士.
略歴1955年奈良県に生る.81年信州大学医学部卒業.81年奈良県立医科大学(福井弘教授)にて小児科の研修,89~92年サンディエゴのScripps研究所留学(Ruggeri教授),93~2009年奈良県立医科大学小児科講師,2009年~血栓制御医学教授.
研究テーマと抱負Von Willebrand因子の構造機能相関,血流を基盤とした血栓形成メカニズムの解明をとおして血栓症の臨床に貢献したい.
趣味スポーツ,映画,読書,旅行.