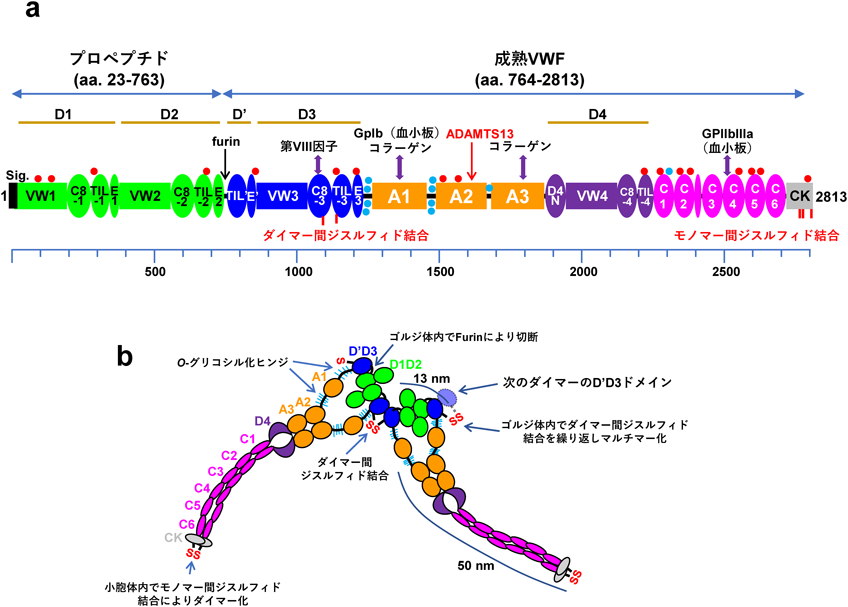
von Willebrand因子(VWF)はヒト血漿中にマルチマーの状態でおよそ10 µg mL−1(モノマー換算で50 nM)1)で存在する巨大糖タンパク質である2).最近の構造学的解析の進展からVWFの従来のドメイン構成が変更され,VWFはN末端側からD1-D2-D′-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CKドメインからなると提唱されている3)(図1a).D1, D2, D3ドメインはVWドメイン,C8フォールド,トリプシンインヒビター様(TIL)構造,Eモジュールの各サブドメインを含む.D′ドメインはVWドメインとC8フォールドを欠き,D4ドメインはEモジュールを欠くがD4Nサブドメインを持つ.血中のVWFは80%以上が血管内皮細胞,残りが巨核球(血小板)由来と考えられている4).小胞体において合成された2813アミノ酸残基のプレプロVWFから22残基のシグナルペプチドが取り除かれ,プロVWFとなる.プロVWFは17か所の高マンノース型N型糖鎖修飾を受ける.VWFの234個のシステイン残基のほとんどは分子内ジスルィド結合を形成するが,C末端側のcysytine knot(CK)ドメイン内の三つのCys残基がVWFモノマー間でジスルフィド結合を形成してダイマー化する(図1b).ゴルジ体に移動したプロVWFは10か所のO型糖鎖が付加されるとともに,複合型糖鎖構造への変換と硫酸化が起こる.VWFの一部の糖鎖にゴルジ体でABO血液型糖鎖が付加される結果,VWFの血中濃度はO型では低くなるなど血液型の影響を受ける5).プロペプチドはfurinによって切断されるが,プロペプチドと2050残基の成熟型VWFの相互作用は非共有結合によって保持される.小胞体よりも酸性で高Ca2+濃度下のゴルジ体内において,VWFダイマーのプロペプチドのD1およびD2ドメインはD3ドメイン内のCys残基のダイマー間ジスルフィド結合の形成を促進し6, 7),超高分子量VWFマルチマー(UL-VWF)が形成される(図1b).UL-VWFは右巻きらせんに組織化されたチューブ状の構造をとり,内皮細胞に特異的な葉巻型の形状の細胞内構造体Weibel-Palade体(WPB)内にコンパクトに蓄えられる8).UL-VWFの分泌は定常状態下でもWPBと細胞膜との融合を介して低レベルで起こっている.血管壁が損傷を受けるなどして周辺の内皮細胞が刺激されると,複数のWPBが集まり分泌ポッドと呼ばれる大きな分泌小胞を形成し,多量のUL-VWFが損傷部位近傍に分泌され止血機能を発揮する.分泌されたVWFはWPBや分泌ポッド内のpH 5.5前後の酸性環境から血中のpH 7.4の環境にさらされると,VWFマルチマーの球状コンホメーションが崩れVWF stringsと呼ばれる長いヒモ状の形状となる.血中でのVWFマルチマーのサイズはおよそ500 kDaから20,000 kDaにわたる.内皮細胞で合成される凝固第VIII因子(FVIII)はVWFと結合した状態でWPBから放出される9).VWFと遊離したFVIIIは速やかにクリアランスされるため,FVIIIの血中安定性にはVWFとの結合が必要である10).VWFの血中半減期はおよそ16時間で,クリアランスには肝実質細胞のアシアロ糖タンパク質受容体(ASPGR-1およびASPGR-2)11)やマクロファージのSIGLEC-512),LRP113),CLEC4M14)などの関与が報告されている.
VWFにはA1-A2-A3ドメインと呼ばれる各200残基程度の3個の連続したドメインが存在し,この領域がVWFの血小板血栓形成能に重要である.Aドメインはいずれもαへリックスで囲まれた六つの並行したβシートを中央に持つRossmannフォールドと呼ばれる構造をとっている15).ジスルフィド結合が多数存在するVWFの他のドメインに比べて三つのAドメイン内にはジスルフィド結合が各ドメインにつき一つしか存在しない.A1およびA3ドメインはN末端とC末端のシステイン残基がジスルフィド結合を形成して比較的しっかりした構造をとる.一方,A2ドメインはC末端の隣り合うシステイン残基(Cys1669およびCys1670)どうしのジスルフィド結合のみで,A1, A3ドメインにはあるα4ヘリックスも存在しないため,構造的に不安定である.細小血管内などの血流が非常に速い部位では血管中央の血流が早い領域と血管壁の遅い領域との間の速度勾配によって生じる物体を歪ませようとする力であるずり応力が高い.このような血流による高ずり応力にさらされると,A2ドメインはアンフォールディングして伸展構造をとる16).
血栓形成においては,初めに血管損傷部位の近傍の内皮細胞からVWFの分泌が促進される.分泌されたVWFはA3ドメイン内のコラーゲン結合部位を介して,血管内皮細胞が剥離して出てきた内皮細胞下組織のコラーゲンに結合する.さらにVWFはA1ドメイン内のGPIbα結合部位を介して血小板表面上の受容体(GPIb-IX)に結合して血小板とも接着する.この相互作用は可逆的で血小板はVWFと接着と乖離を繰り返す(ローリング).血小板はGPVIなどのコラーゲン受容体を介して直接内皮下組織にも結合する.ローリングの間のGPIb-IXとVWFの相互作用から生じるinside-outシグナルにより血小板のインテグリンαIIbβ3(GPIIb/IIIa)が活性化される.その結果血小板のαIIbβ3はVWFのC1ドメイン上のRGD配列を介してVWFに結合し強固な血小板粘着が起こる.VWFと結合したαIIbβ3からoutside-inシグナルが入ると,血小板内のCa2+濃度が上昇してα顆粒内のフィブリノゲンやVWF,濃染顆粒内のADPやセロトニンの開口分泌が促進される.放出されたADPはADP受容体を介してトロンボキサンA2の活性化を促し,周囲の血小板を二次的に活性化する.活性化された血小板上のαIIbβ3はVWFやフィブリノゲンと血小板を架橋し血小板を凝集させる.同時に血小板の扁平化と伸展が進み,最終的に血小板凝集塊が形成される.(一次止血).一部の活性化血小板では細胞膜の内側からホスファチジルセリンなどの酸性リン脂質が表面に出現する.その結果,血小板上に多くの凝固因子が集まって凝固系が活性化しトロンビンが産生され,フィブリノゲンをフィブリンに転換し強固な不溶性フィブリン網を形成して最終的な止血に至る(二次止血).
2. von Willebrand病(von Willebrand disease:VWD)
以上みてきたように,VWFは一次止血に関わる血小板血栓形成において重要な役割を果たしている.したがってVWFの遺伝子の変異による質的・量的欠損は先天性の出血障害VWDの病因となる17).VWD患者では粘膜出血や皮下出血,手術および他の止血負荷に伴う出血が起こる.軽度のVWDでは明らかな症状が軽度の止血困難だけであるため気づかれないことも多く,その潜在数も1万人あたり1~100人と幅がある.VWDはVWF生成の量的欠損(1型),質的異常(2型),完全欠損(3型)に分類される.1型VWDは全体の70%程度を占める.全体の20~30%を占める2型VWDはUL-VWFの欠損がみられる2A型,VWFと血小板の親和性が亢進してUL-VWFが減少する2B型,UL-VWFは存在するものの,その機能異常により止血異常を示す2M型,FVIIIとの親和性が低下して止血異常を示す2N型に分類される.また頻度は5~10%程度と低いものの,3型VWDはVWFの完全欠損により重篤な出血症状を示す.VWDを引き起こす遺伝子変異は遺伝子欠失,ストップコドンの挿入,フレームシフト,スプライシング部位変異,プロモーター内変異などさまざまである.1型および2型VWDにおいては変異型のVWFが正常型VWFとともに同じマルチマーに取り込まれてドミナントネガティブに作用し,全体のマルチマーの量や活性に影響する結果,常染色体優性の表現型を示すことが多い.3型VWDは変異をホモもしくは複合ヘテロで持つために起こるVWFの完全欠損で劣性遺伝形式である.治療はVWFの補充のためにVWFを含む第VIII因子濃縮製剤の輸注が行われる.バゾプレシンのアナログであるデスモプレシン酢酸塩水和物(DDAVP)は内皮細胞からのVWFの放出を促進するために1型VWDの治療に用いられる.
3. 後天性von Willebrand症候群(acquired von Willebrand syndrome:AVWS)
VWF遺伝子の変異を伴わず後天的にVWD様の病態を示す疾患をAVWSと総称する18).AVWSの多くは出血症の既往がない高年齢層で他の基礎疾患を背景に生じる.AVWSは以下のような要因で血中VWF量もしくはVWFマルチマーサイズが減少するために起こると考えられている.(1)全身性エリテマトーデスや高リン脂質抗体症候群などの自己免疫疾患等における,VWFに対する自己抗体の出現による血中からのVWFのクリアランスの亢進.(2)非血液性腫瘍,リンパ増殖性疾患,骨髄増殖性疾患等における腫瘍細胞表面へのVWFの接着と取り込み.(3)甲状腺機能低下症におけるVWF合成の低下.(4)バルプロ酸などの特定の薬剤投与.(5)重症大動脈弁狭窄症の狭窄部位や末期心疾患患者の心臓機能の補助に用いられる左室補助循環装置(LVAD)ポンプ内の高ずり応力下におけるADAMTS13によるVWFの切断(次項参照)19).治療にはAVWSの多様な発症機序に合わせて,デスモプレシン,VWF含有凝固因子製剤,免疫グロブリン,免疫抑制剤などが用いられる.
LVAD装着例のほぼすべてでVWFマルチマーサイズが減少すると考えられるが,臨床現場でVWFマルチマーサイズを測定することは困難でAVWSと確定診断される症例は少ない.AVWSの多くで消化管出血が起こり治療に支障をきたす.LVADポンプの駆動形式や設定がVWFマルチマーサイズへ及ぼす影響やVWFマルチマーサイズと臨床症状との相関を明らかにすることは診断や治療にとって重要であり,研究の進展が期待される.
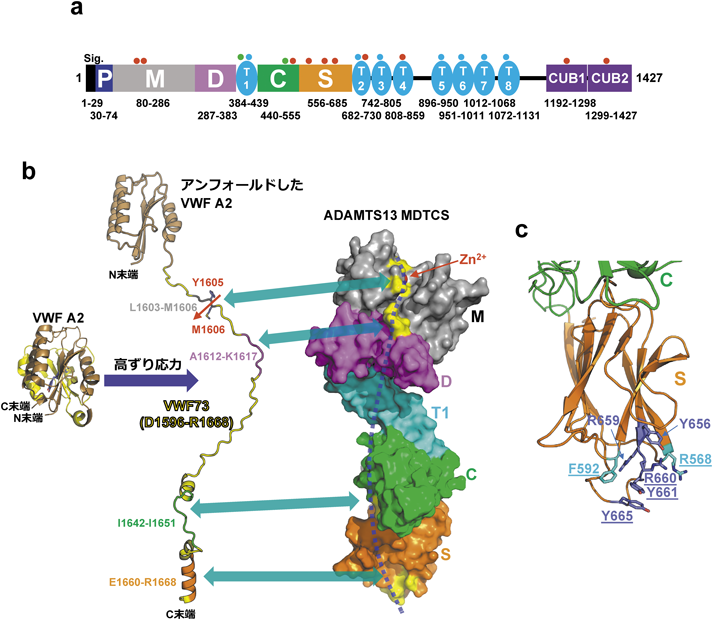
UL-VWFの蓄積は望ましくない過剰な血小板凝集をもたらす.マルチドメインからなる分泌型メタロプロテアーゼADAMTS13(a disintegrin-like and metalloprotease with thrombospondin type 1 motif-13)はずり応力依存的にVWF A2ドメイン内のTyr1605-Met1606間を特異的に切断しUL-VWFを適度なレベルに保つことで過剰な血小板凝集を防いでいる20).ADAMTS13は主に肝臓の星細胞で合成され21),血中へ分泌される.その血中濃度は0.7~1.4 µg mL−1(3.5~7.0 nM)である22).ADAMTS13はヒトでは19の遺伝子からなるADAMTSファミリーに属するメタロプロテアーゼで,N末端側のメタロプロテアーゼ(M)ドメインの後ろに,ディスインテグリン様(D),トロンボスポンジン-1(T1),システインリッチ(C),スペーサー(S)ドメインが続く(図2a).Mドメインおよびその隣接領域(MDTCS)はすべてのADAMTSプロテアーゼに存在し,各プロテアーゼの特異的な活性に関わっていると考えられている.in vitroでのADAMTS13によるVWFの切断にはMDTCSが必要かつ十分である.ADAMTS13ではMDTCSのC末端側に七つのトロンボスポンジン-1(T2~T8)と二つのCUB(complement C1r/C1s, Uegf, Bmp1)ドメイン(CUB1, CUB2)が並ぶ.
ADAMTS13の切断反応はVWF A2ドメインの立体構造に依存して調節されている.DTCSの構造とMDTCSモデル23)および変異体の切断活性などをもとにして,MDTCSによる切断は以下のステップで起こると考えられている.(1)定常条件下ではVWF A2ドメイン内のADAMTS13切断部位は球状構造をとるドメインの内部に埋もれている(図2b).(2)前述したようにVWFのA2ドメインは高ずり応力がかかると球状構造から伸展構造に変化する.(3)伸展したA2ドメインのC末端側領域(aa. 1596-1668, VWF73)はADAMTS13の空間的に隔たりながらも直線状に並んだD, C, S各ドメイン上に存在する複数のVWF結合エキソサイトへ結合する.(4)その結果,Mドメインの活性部位でTyr1605-Met1606間を特異的に切断する(図2b).血栓が成長すると血管内腔が狭くなってずり応力が高まり,VWFを介した血小板の集積・血栓の成長が促進される.一方でADAMTS13によるVWFの切断も促進され,血管が血栓の成長で閉塞してしまわないように調節していると考えることができる.実際に内皮細胞表面のVWF stringsの形成と伸長はVWF stringsのADAMTS13よる切断によって厳密に調節されていることが知られている24).
ADAMTS13活性がADAMTS13遺伝子の変異もしくはADAMTS13タンパク質に対する中和抗体の出現により3%以下に著減すると,血中にUL-VWFが蓄積し細小血管内で過剰な血小板凝集が起こる.全身で微小血小板血栓が形成されると,血小板の消費による血小板減少症,血栓をすり抜けようとする赤血球が機械的に破壊されて溶血性貧血,腎臓の毛細血管が血栓で閉塞することで腎機能障害が引き起こされる.これらを主な病状とする血栓性疾患が血栓性血小板減少性紫斑病(thrombotic thrombocytopenic purpura:TTP)である25).これらの症状は溶血性尿毒症症候群(hemolytic uremic syndrome:HUS)と共通している.TTPとHUSの鑑別は臨床的には困難であることが多いため,近年は血栓性微小血管障害症(thrombotic microangiopathy:TMA)という共通の病理学的診断名が用いられている.TTPとHUSの識別にはADAMTS13のVWF切断活性の測定が必要である.TTPはきわめてまれな疾患(人口100万人あたり毎年4人程度)だが,治療しない場合の致死率は非常に高い.TTPの9割以上はADAMTS13に対する中和抗体(インヒビター)の出現によるADAMTS13活性の阻害により起こる後天性TTPである.ADAMTS13の遺伝子変異によって引き起こされる先天性TTPはUpshaw-Schulman症候群(USS)とも呼ばれる.USSの遺伝子変異はVWD同様にさまざまでADAMTS13遺伝子の全領域にわたる.コード領域に変異を持つADAMTS13はほぼすべてが分泌異常によって活性が著減すると考えられている.USSは常染色体劣性で患者は変異を両親から受け継ぎ複合型ヘテロで発症する場合が多い.TTPの治療にはADAMTS13を補充するための血漿交換療法が行われている26).TTP患者に輸注された新鮮凍結血漿中のADAMTS13活性の半減期は2~3日である27).ごく最近,ADAMTS13のクリアランスにマクロファージのCD163が関与することが明らかになった28).後天性TTPのうち血漿交換療法に治療抵抗性を示し高いインヒビター価を示す症例で,抗ヒトCD20ヒト・マウスキメラのモノクローナル抗体製剤のリツキシマブが有効であることが報告されている29).また,抗VWFヒト化単一可変領域免疫グロブリン(ナノボディ)製剤のカプラシズマブは血小板とUL-VWFとの結合を阻害することで,後天性TTPの病態を改善すると期待され治験が進行中である30).
ADAMTS13の活性阻害抗体を引き起こす中和抗体のエピトープ部位のほとんどが,ADAMTS13のSドメインの先端の複数のループに存在するVWF結合エキソサイト31, 32)(図2c)およびその近傍であることが明らかにされた.このループに複数の変異(F592Y/R568K/R660K/Y661F/Y665F,図2c)を導入したADAMTS13は,ほとんどの中和抗体に対しての親和性が減弱し,野生型に比べて中和抗体による活性阻害を受けにくくなることが示された33).これらの結果から,後天性TTPにおいては中和抗体によってADAMTS13のSドメイン先端ループ内のVWF結合エキソサイトへのVWFの結合が妨げられ,ADAMTS13のVWF切断が阻害されていると推測されている.
6. ADAMTS13のコンホメーション変化による活性調節
前述のようにVWFの切断にはADAMTS13のN末端側MDTCSと高ずり応力下でアンフォールディングしたVWF A2ドメインのC末端側領域VWF73との相互作用が必要である.一方ADAMTS13のC末端側T5-CUB2はずり応力非依存的にVWF D4-CKドメインに可逆的に結合する34, 35).ADAMTS13のC末端側T2-CUB2の欠失はin vitroでのペプチド基質の切断を増強させVWFマルチマー切断を減弱させる36)が,ADAMTS13のC末端側ドメインのADAMST13活性調節機構は最近まで不明だった.SadlerらのグループはADAMTS13のVWF切断活性を増加させるADAMTS13のT6-CUB2に対する抗体を見いだし,この抗体の結合により,ADAMTS13が活性型のコンホメーションをとるものと考えた37).彼らはADAMTS13のペプチド基質VWF71の切断活性がpH 6.0においてpH 7.4に比べておよそ8倍高いこと,反応液中へのVWF D4ドメインの添加が活性を増強することなどから,VWFのC末端側D4-CKにADAMTS13のC末端側領域が結合することでADAMTS13がアロステリックに活性化されると推測した.ADAMTS13のX線小角散乱データをMDTCS構造モデルに当てはめてADAMTS13の全長構造を推定した結果,ADAMTS13のC末端側領域は折れ曲がり,C末端の二つのCUBドメインがADAMTS13のSドメイン領域に覆いかぶさる構造をとっていると推測された.LaneらのグループもSドメインがCUB1-2ドメインと相互作用すること,透過型電子顕微鏡による全長ADAMTS13分子の観察から,ADAMTS13が開いたコンホメーションと折りたたまれたコンホメーションをとっていることを示した38).さらに前述のSドメインループの変異(F592Y/R568K/R660K/Y661F/Y665F,図2c)のADAMTS13は野生型よりより開いたコンホメーションをとりやすいことを明らかにした.
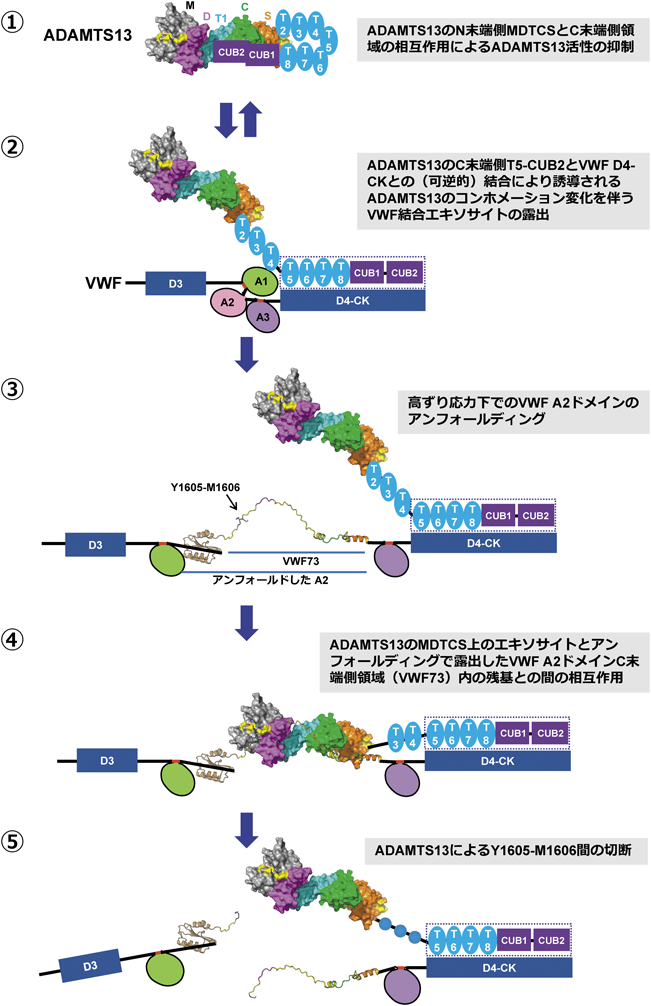
以上の結果から図3のようなADAMTS13によるVWF切断機構が提唱されている.(1)折りたたまれたコンホメーションのADAMTS13はMDTCSの活性部位とVWF結合エキソサイトがC末端側領域によって覆い隠され活性が低い,(2)一方,VWFの存在下ではADAMTS13のT5-CUB2がVWFのD4-CKへ結合し,ADAMTS13は開いたコンホメーションに転換する,(4)高ずり応力下でVWF A2ドメインがアンフォールディングする,(5)ADAMTS13のMDTCSがアンフォールディングしたA2ドメインに結合してVWFを切断する(図3).ADAMTS13はVWFが存在しないときには閉じたコンホメーションをとり凝固因子などの血中プロテアーゼによる分解を防いでいる可能性が考えらえる.VWFとの相互作用によるADAMTS13のコンホメーション変化を伴う活性化は,ずり応力によるVWF A2ドメインのアンフォールディング依存的にMDTCSが複数のエキソサイトを介してVWFを認識する機構とともに高濃度の血漿タンパク質(70~80 mg mL−1)中の微量VWF(10 µg mL−1)をADAMTS13が特異的に効率よく切断することを可能にしている.一方で,ADAMTS13の開いたコンホメーションへの転換は隠れていたSドメイン先端のループを表面へ露出させることで免疫系に異物認識され中和抗体の産生を誘導し後天性TTPの発症を引き起こす原因にもなっている可能性がある.
最近,VWFと結合するGPIbαに60アミノ酸残基からなる機械受容ドメイン(MSD)が同定された39).高ずり応力下でVWF A1ドメインがGPIbαのMSDに結合するとMSDがアンフォールディングして複合体をともに構成しているGPIbβとGPIXのコンホメーション変化を引き起こし,血小板内のシグナル伝達系が惹起される.ずり応力依存的なアンフォールディングによる調節機構がリガンド(VWF)側にも存在することは興味深い.静止状態とは異なる生体を反映したずり応力下での血栓形成機構がin vitroにおける分子ピンセット,結晶構造解析,X線小角散乱,電子顕微鏡を用いた単粒子解析,フローチャンバーシステム,in vivoにおけるノックアウトマウスを用いた生体分子イメージングによる血栓形成の観察など多様な実験手法の進展により明らかとなってきている.今後のさらなる研究の進展が期待される.
ADAMTS13活性の低下によって起こるUL-VWFの蓄積は心筋梗塞40)・虚血性脳梗塞41),妊娠高血圧症候群42),重篤な脳マラリア43)のリスクと相関がある.またマウスなどの動物モデルを用いた実験からADAMTS13ノックアウトマウスでは心臓や脳での虚血再灌流後の障害が大きくなるなど,ADAMTS13はさまざまな動脈血栓性疾患へ関与している可能性がある42).高脂肪食を与えたAdamts13ノックアウトマウスは大動脈動脈硬化巣のプラークの数と大きさが増大し,組織内へのマクロファージの浸潤が増加する45, 46)が,この影響はVWF依存性である47).ADAMTS13-VWF系が血栓形成機能を超えて炎症反応にも関与している可能性が示唆される.ADAMTS13-VWF系のさらなる研究が循環器疾患の病態の解明と治療への応用に役立っていくことが期待される.
引用文献References
1) Borchiellini, A., Fijnvandraat, K., ten Cate, J.W., Pajkrt, D., van Deventer, S.J., Pasterkamp, G., Meijer-Huizinga, F., Zwart-Huinink, L., Voorberg, J., & van Mourik, J.A. (1996) Blood, 88, 2951–2958.
2) Springer, T.A. (2014) Blood, 124, 1412–1425.
3) Zhou, Y.-F., Eng, E.T., Zhu, J., Lu, C., Walz, T., & Springer, T.A. (2012) Blood, 120, 449–458.
4) McGrath, R.T., McRae, E., Smith, O.P., & O’Donnell, J.S. (2010) Br. J. Haematol., 148, 834–843.
5) Franchini, M., Crestani, S., Frattini, F., Sissa, C., & Bonfanti, C. (2014) Clin. Chem. Lab. Med., 52, 1273–1276.
6) Mayadas, T.N. & Wagner, D.D. (1992) Proc. Natl. Acad. Sci. USA, 89, 3531–3535.
7) Haberichter, S.L. (2015) Blood, 126, 1753–1761.
8) Zhou, Y.-F., Eng, E.T., Nishida, N., Lu, C., Walz, T., & Springer, T.A. (2011) EMBO J., 30, 4098–4111.
9) Turner, N.A. & Moake, J.L. (2015) PLoS One, 10, e0140740.
10) Lenting, P.J., VAN Schooten, C.J.M., & Denis, C.V. (2007) J. Thromb. Haemost., 5, 1353–1360.
11) Grewal, P.K., Uchiyama, S., Ditto, D., Varki, N., Le, D.T., Nizet, V., & Marth, J.D. (2008) Nat. Med., 14, 648–655.
12) Pegon, J.N., Kurdi, M., Casari, C., Odouard, S., Denis, C.V., Christophe, O.D., & Lenting, P.J. (2012) Haematologica, 97, 1855–1863.
13) Rastegarlari, G., Pegon, J.N., Casari, C., Odouard, S., Navarrete, A.-M., Saint-Lu, N., van Vlijmen, B.J., Legendre, P., Christophe, O.D., Denis, C.V., & Lenting, P.J. (2012) Blood, 119, 2126–2134.
14) Rydz, N., Swystun, L.L., Notley, C., Paterson, A.D., Riches, J.J., Sponagle, K., Boonyawat, B., Montgomery, R.R., James, P.D., & Lillicrap, D. (2013) Blood, 121, 5228–5237.
15) Huizinga, E.G., Martijn van der Plas, R., Kroon, J., Sixma, J.J., & Gros, P. (1997) Structure, 5, 1147–1156.
16) Zhang, X., Halvorsen, K., Zhang, C.-Z., Wong, W.P., & Springer, T.A. (2009) Science, 324, 1330–1334.
17) Leebeek, F.W.G. & Eikenboom, J.C.J. (2016) N. Engl. J. Med., 375, 2067–2080.
18) Mital, A. (2016) Adv. Clin. Exp. Med., 25, 1337–1344.
19) Nascimbene, A., Neelamegham, S., Frazier, O.H., Moake, J.L., & Dong, J.F. (2016) Bood, 127, 3133–3141.
20) Zheng, X.L. (2015) Annu. Rev. Med., 66, 211–225.
21) Uemura, M., Tatsumi, K., Matsumoto, M., Fujimoto, M., Matsuyama, T., Ishikawa, M., Iwamoto, T.-A., Mori, T., Wanaka, A., Fukui, H., & Fujimura, Y. (2005) Blood, 106, 922–924.
22) Rieger, M., Ferrari, S., Kremer Hovinga, J.A., Konetschny, C., Herzog, A., Koller, L., Weber, A., Remuzzi, G., Dockal, M., Plaimauer, B., & Scheiflinger, F. (2006) Thromb. Haemost., 95, 212–220.
23) Akiyama, M., Takeda, S., Kokame, K., Takagi, J., & Miyata, T. (2009) Proc. Natl. Acad. Sci. USA, 106, 19274–19279.
24) Dong, J., Moake, J.L., Nolasco, L., Bernardo, A., Arceneaux, W., Shrimpton, C.N., Schade, A.J., McIntire, L.V., Fujikawa, K., & López, J.A. (2002) Blood, 100, 4033–4039.
25) Rogers, H.J., Allen, C., & Lichtin, A.E. (2016) Cleve. Clin. J. Med., 83, 597–603.
26) Scully, M. & Blombery, P. (2014) J. Blood Med., 5, 15.
27) Furlan, M., Robles, R., Morselli, B., Sandoz, P., & Lämmle, B. (1999) Thromb. Haemost., 81, 8–13.
28) Verbij, F.C., Sorvillo, N., Kaijen, P.H.P., Hrdinova, J., Peyron, I., Fijnheer, R., Brinke, A., Meijer, A.B., Van Alphen, F.P.J., Van Den Berg, T.K., Graversen, J.J.H., Moestrup, S.K., & Voorberg, J. (2017) Blood Adv., 1, 8–12.
29) Page, E.E., Kremer Hovinga, J.A., Terrell, D.R., Vesely, S.K., & George, J.N. (2016) Blood, 127, 3092–3094.
30) Peyvandi, F., Scully, M., Kremer Hovinga, J.A., Cataland, S., Knöbl, P., Wu, H., Artoni, A., Westwood, J.-P., Mansouri Taleghani, M., Jilma, B., Callewaert, F., Ulrichts, H., Duby, C., & Tersago, D., & TITAN Investigators. (2016) N. Engl. J. Med., 374, 511–522.
31) Pos, W., Crawley, J.T.B., Fijnheer, R., Voorberg, J., Lane, D.A., & Luken, B.M. (2010) Blood, 115, 1640–1649.
32) Jin, S.-Y., Skipwith, C.G., & Zheng, X.L. (2010) Blood, 115, 2300–2310.
33) Jian, C., Xiao, J., Gong, L., Skipwith, C.G., Jin, S.-Y., Kwaan, H.C., & Zheng, X.L. (2012) Blood, 119, 3836–3843.
34) Feys, H.B., Anderson, P.J., Vanhoorelbeke, K., Majerus, E.M., & Sadler, J.E. (2009) J. Thromb. Haemost., 7, 2088–2095.
35) Zanardelli, S., Chion, A.C.K., Groot, E., Lenting, P.J., McKinnon, T.A.J., Laffan, M.A., Tseng, M., & Lane, D.A. (2009) Blood, 114, 2819–2828.
36) Zhang, P., Pan, W., Rux, A.H., Sachais, B.S., & Zheng, X.L. (2007) Blood, 110, 1887–1894.
37) Muia, J., Zhu, J., Gupta, G., Haberichter, S.L., Friedman, K.D., Feys, H.B., Deforche, L., Vanhoorelbeke, K., Westfield, L.A., Roth, R., Tolia, N.H., Heuser, J.E., & Sadler, J.E. (2014) Proc. Natl. Acad. Sci. USA, 111, 18584–18589.
38) South, K., Luken, B.M., Crawley, J.T.B., Phillips, R., Thomas, M., Collins, R.F., Deforche, L., Vanhoorelbeke, K., & Lane, D.A. (2014) Proc. Natl. Acad. Sci. USA, 111, 18578–18583.
39) Zhang, W., Deng, W., Zhou, L., Xu, Y., Yang, W., Liang, X., Wang, Y., Kulman, J.D., Zhang, X.F., & Li, R. (2015) Blood, 125, 562–569.
40) Matsukawa, M., Kaikita, K., Soejima, K., Fuchigami, S., Nakamura, Y., Honda, T., Tsujita, K., Nagayoshi, Y., Kojima, S., Shimomura, H., Sugiyama, S., Fujimoto, K., Yoshimura, M., Nakagaki, T., & Ogawa, H. (2007) Am. J. Cardiol., 100, 758–763.
41) Hanson, E., Jood, K., Nilsson, S., Blomstrand, C., & Jern, C. (2009) J. Thromb. Haemost., 7, 2147–2148.
42) Stepanian, A., Cohen-Moatti, M., Sanglier, T., Legendre, P., Ameziane, N., Tsatsaris, V., Mandelbrot, L., de Prost, D., & Veyradier, A., & ECLAXIR Study Group. (2011) Arterioscler. Thromb. Vasc. Biol., 31, 1703–1709.
43) Larkin, D., de Laat, B., Jenkins, P.V., Bunn, J., Craig, A.G., Terraube, V., Preston, R.J.S., Donkor, C., Grau, G.E., van Mourik, J.A., & O’Donnell, J.S. (2009) PLoS Pathog., 5, e1000349.
44) Fujioka, M., Hayakawa, K., Mishima, K., Kunizawa, A., Irie, K., Higuchi, S., Nakano, T., Muroi, C., Fukushima, H., Sugimoto, M., Banno, F., Kokame, K., Miyata, T., Fujiwara, M., Okuchi, K., & Nishio, K. (2010) Blood, 115, 1650–1653.
45) Gandhi, C., Khan, M.M., Lentz, S.R., & Chauhan, A.K. (2012) Blood, 119, 2385–2391.
46) Jin, S.-Y., Tohyama, J., Bauer, R.C., Cao, N.N., Rader, D.J., & Zheng, X.L. (2012) Arterioscler. Thromb. Vasc. Biol., 32, 1817–1823.
47) Gandhi, C., Ahmad, A., Wilson, K.M., & Chauhan, A.K. (2014) J. Thromb. Haemost., 12, 255–260.
著者紹介Author Profile
 秋山 正志(あきやま まさし)
秋山 正志(あきやま まさし)国立循環器病研究センター研究所分子病態部室長.理学博士.
略歴1996年岡山大学大学院自然科学研究科(博士課程)修了,96年早稲田大学助手,2006年国立循環器病センター研究員,09年から現職.
研究テーマと抱負ADAMTS13はTTPのみならず様々な血栓症の病態の改善につながる可能性を持っている.多数の方が罹患する血栓症の理解と病態の改善につながる研究を目指したい.
ウェブサイトhttp://www.ncvc.go.jp/res/divisions/etiology/
趣味読書.