発作性夜間ヘモグロビン尿症(paroxysmal nocturnal hemoglobinuria,以下PNHと略記する)は,PIGA遺伝子の体細胞変異によってグリコシルホスファチジルイノシトール(glycosylphosphatidylinositol,以下GPIと略記する)アンカー生合成が欠損した多能性造血幹細胞ができ,それが骨髄不全を起こした人においてクローン拡大を起こし,GPIアンカー型タンパク質を欠損した異常血液細胞を大量に作ることによって発症する.GPIアンカー型の補体制御因子を欠損した赤血球は,自己補体に対する抵抗性を失い,感染などに伴って活性化する補体によって破壊される.本稿では,PNHの疾患概念,GPIアンカー型タンパク質の生合成,補体制御における役割,PNHの生化学的・分子遺伝学的異常について概説し,最後に治療についてもふれる.
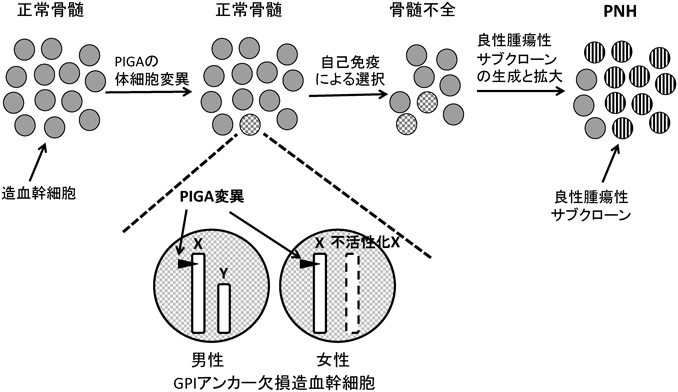
PNHは後天性の難治性血液疾患で,発症時期は小児期から高齢者まで幅広いが,40歳代から60歳代が中心であり,小児例は少ない1–3).患者数は,人口10万人に1~2人あるいはそれ以下である1).わが国では指定難病であり,患者数は1000人以上と推定される.造血幹細胞に異常があるため,発症すると10年以上,25年さらに30年と長期に持続する.PNHの3主徴は,血管内溶血,血栓と骨髄不全である.補体による溶血と骨髄不全で起こる貧血,遊離ヘモグロビンによる一酸化窒素の除去に伴う諸症状(嚥下困難,肺高血圧,勃起不全等)と腎臓への負荷,輸血に伴う諸症状等が多くの患者のQOLを大きく低下させる.血栓は肝静脈など深部静脈を中心に各所で起こることが特徴で,わが国の患者では比較的少ないが,欧米の患者では最大の死因である.血栓は,溶血に伴って起こることが多く,補体の活性化が関わっている.異常造血幹細胞由来の異常血小板の関与が考えられるが,メカニズムの詳細は不明である.骨髄不全は,補体制御因子欠損とは別に,自己免疫機序で起こる.細胞障害により正常造血幹細胞数を減少させることで異常幹細胞クローンの拡大に寄与していると考えられる.
PNH患者の血液には,自己補体に感受性が高い異常赤血球のクローン性の集団が出現していて,異常赤血球と正常赤血球が共存した状態になっている.異常赤血球の割合は,10%程度から90%に達するまで症例間で幅広く違いがある.異常赤血球では,補体から自己細胞を保護する細胞膜上の補体制御因子であるDAF(decay-accelerating factor)/CD55とCD59が完全欠損しているか,あるいは大きく低下している.DAFとCD59を欠損した異常細胞集団は,血小板,好中球,単球,T,B,NKリンパ球にも現れる.
DAFは,自己細胞上にC4b2aやC3bBbといったC3転換酵素ができてしまったときに,速やかに触媒サブユニットであるC2aあるいはBbをC4bあるいはC3bから解離させることにより失活させる.CD59は,自己細胞に結合したC5b-7がC5b-8になった段階で結合し,C9の結合を阻害することにより,膜障害性複合体が完成するのを防ぐ.正常赤血球は,DAFとCD59を発現しており,補体経路の中心酵素であるC3転換酵素形成のステップと膜障害性複合体形成のステップの2か所を阻害する機能により,自己補体に一定の抵抗性を持っている.PNHの異常赤血球は,DAFとCD59の欠損により,こうした制御メカニズムが働かないため,補体の活性化に伴って膜障害性複合体形成が起こり,溶血する.
補体は異物排除のエフェクターとして自然免疫の重要な要素であるが,系に内在する自己細胞障害のリスクがある.一つは,C4とC3が反応性の分子内チオエステルを持っていて,活性化で生じるC4bとC3bが異物にエステルあるいはアミド結合で共有結合する仕組みであるが,必然的に近傍の自己細胞表面にも一定の割合で結合してしまうというリスクである.また,後期経路でできるC5b-7複合体には脂質結合部位ができ,脂質膜に結合する.その後C8と複数のC9が結合すると膜障害作用を持つMAC(membrane attack complex)となり,微生物の表面膜を破壊するが,C5b-7は近傍の自己細胞表面に結合するリスクがある.これらの自己細胞障害リスクは,自己細胞上に補体制御因子を持つことで回避されている.
補体は,感染などに伴って活性化するので,PNHの異常赤血球は,それによって一斉に破壊され溶血発作を呈する.また,補体の第二経路は常に少しずつ活性化しており,その結果PNHの異常赤血球は常に少しずつ溶血している.機序は不明であるが,睡眠中には活性化レベルが上がるらしく,溶血が亢進する.こうした特徴から,PNHの病名がつけられた.
好中球などの有核細胞では,膜貫通型の補体制御因子であるCD46が発現しており,さらにMACを除去(たとえばエンドサイトーシスなどによって)したり,修復したりする機能が備わっており,DAFとCD59を欠損しても補体抵抗性を持っているため,破壊を免れる.
DAFとCD59はともにGPIアンカー型タンパク質であり,PNHの異常はGPIアンカーの生合成欠損であるので,次にGPIアンカー型タンパク質を概説する.
GPIアンカー型タンパク質は膜内在性タンパク質であるが,タンパク質部分自体は直接膜に挿入されていない.代わりに,カルボキシ末端(C末端)にアミド結合した糖脂質であるGPIの脂肪鎖(脂肪酸あるいは脂肪族アルコール)が,細胞膜の外葉に挿入され,タンパク質を細胞膜上に係留している(図1).
GPIアンカーは,真核生物に広く用いられている膜アンカー構造で,単細胞生物である原生動物では細胞表面の主要タンパク質の多くがGPIアンカー型である.出芽酵母では,およそ6000遺伝子のうち少なくとも60がGPIアンカー型タンパク質の遺伝子である.ヒトでは150種以上のタンパク質がGPIアンカー型タンパク質である4).新たなタンパク質が見つかってきており,リストはさらに大きくなりつつある5).酵素(アルカリホスファターゼ,5′-ヌクレオチダーゼ等),受容体(葉酸受容体,Fcγ受容体IIIB等),接着因子(contactin, CD58等),補体制御因子(CD59, DAF/CD55),プリオンタンパク質など,機能的にも構造的にもさまざまなタンパク質がGPIアンカー型である.
糖脂質であるGPIアンカーの構造は,糖鎖部分の骨格と側鎖,そして脂質部分に分けることができる.骨格は,生物種を問わず保存された構造で,EtNP-6Manα1,2Man-α1,6Man-α1,4GlcNα1,6myoInositolphosphate(EtNP:エタノールアミンリン酸,Man:マンノース,GlcN:グルコサミン)である.エタノールアミンのアミノ基がタンパク質C末端とアミド結合している.N-アセチル化されていないグルコサミンが最終構造として存在することがGPIアンカー糖鎖の特徴である.グルコサミンに結合しているマンノースから順に第1,第2,第3マンノースと呼ぶ.
側鎖は,生物種やタンパク質によって特徴があり,骨格の三つのマンノースにさまざまな糖鎖,あるいはエタノールアミンリン酸が結合していることが知られている.哺乳動物では,第1マンノースの2位にエタノールアミンリン酸が必ず側鎖として結合している.このエタノールアミンリン酸側鎖は,GPIアンカー型タンパク質の発現に重要で,欠損すると発現が低下する.また,同じ第1マンノースの4位にN-アセチルガラクトサミン(GalNAc)がβ結合している場合があり,GalNAcにはβ1,3ガラクトース,さらにシアル酸が結合し,最長3糖の側鎖になる.また一方,第4マンノースが,側鎖として第3マンノースにα1,2結合している場合もある.これらの糖側鎖の生理的意義はまだ不明である.
脂質部分も生物種やタンパク質によって違いがあり,ジアシル型あるいは1-アルキル-2-アシル型ホスファチジルイノシトール(PI)または,イノシトールホスホセラミドである.脂質部分は,生合成の出発材料である小胞体のPIがそのまま成熟したタンパク質GPIアンカーに残っているのではなく,生合成過程でリモデリングを受け,元の構造とは大きく変化している.たとえば,原生動物である睡眠病トリパノソーマの表面を覆っているvariant surface glycoproteinと呼ばれるGPIアンカー型タンパク質は,ジアシルグリセロール型PIのsn-1, sn-2両方の脂肪酸が小胞体においてミリスチン酸に置き換えられる脂肪酸リモデリングを受けてからタンパク質に付加される.出芽酵母では,タンパク質によってPIがイノシトールホスホセラミドに変換される.PI型であるタンパク質においても,sn-2の脂肪酸は,飽和型C26という極長鎖脂肪酸に置き換えられている4).
ヒトのGPIアンカー型タンパク質は,1-アルキル-2-アシル型PIが主体で,ジアシル型PIは少数成分である.これらの構造も,出発材料のPIとは大きく異なっており,小胞体での生合成過程においてジアシル型から1-アルキル-2-アシル型に変化するリモデリングと,さらにゴルジ体においてsn-2の脂肪酸がステアリン酸に置換する脂肪酸リモデリングを経て形成される.脂肪酸リモデリングによってGPIアンカー型タンパク質は2本の飽和脂肪鎖を持ち,脂質ラフトへ限局する.1-アルキル型PIの生理的意義はまだ明らかではない4).
5. GPIアンカー型タンパク質の生合成と生合成遺伝子群
GPIアンカー型タンパク質は,小胞体で生合成されたGPIアンカー前駆体が,CD59などの前駆タンパク質のC末端に翻訳後修飾されてできる(図1).前駆タンパク質はC末端にGPI付加シグナル配列を持ち,シグナルペプチドとGPIが置き換わることによりGPI付加が行われる4, 6).
小胞体でのGPIアンカー前駆体の生合成は,11のステップを経て行われ,少なくとも17遺伝子が関わっている(図1).生合成は,細胞質側にあるPIにUDP-N-アセチルグルコサミン(UDP-GlcNAc)から,GlcNAcが転移される反応で始まる.このステップを触媒するGPI-N-アセチルグルコサミン転移酵素は,七つのタンパク質(PIGA, PIGC, PIGH, PIGP, PIGQ, PIGY, DPM2)からできた複合体酵素である7, 8).次いで,脱アセチル酵素(PIGL)によってGlcN-PIに変化し,それが小胞体の内腔側にフリップし,その後の反応は内腔側で進行する.フリップのメカニズムは未解決である.小胞体内腔では,まずGlcN-PIのイノシトールの2位にパルミチン酸がアシル転移酵素(PIGW)によって付加され,次にPI部分がジアシル型から1-アルキル-2-アシル型が主成分に変化する.この脂質リモデリングの詳細は未解決であるが,1-アルキル-2-アシル型を主成分とするリン脂質(ホスファチジルエタノールアミンは一つの候補である)とGlcN-(inositol-acylated)PIの間でジラジルグリセロールあるいはジラジルホスファチジン酸の交換が行われるのであろうと推定されている.1-アルキルグリセロール部分の合成はペルオキシソーム依存性である.その後,三つのマンノースと三つのエタノールアミンリン酸が順に付加され,GPIアンカー前駆体が完成する.すべてのマンノースは,ドリコールリン酸マンノースから,すべてのエタノールアミンリン酸は,ホスファチジルエタノールアミンから供給される.第1,第2,第3マンノースを転移する三つのマンノース転移酵素であるPIGM, PIGV, PIGBは,いずれも複数膜貫通型の小胞体膜タンパク質である.このうちPIGMは,1回膜貫通型タンパク質であるPIGXと複合体を作ることによって安定化されている.第1マンノースの2位に側鎖として結合するエタノールアミンリン酸は,PIGNによって転移され,第3マンノースの6位に結合しアンカーをタンパク質につなげるエタノールアミンリン酸は,PIGO/PIGF複合体によって転移される.また,第2マンノースの6位に結合するエタノールアミンリン酸は,PIGG/PIGF複合体によって,生合成の最後に転移される.このエタノールアミンリン酸は,GPIがタンパク質に付加された後,小胞体から輸送小胞へ取り込まれるまでに除去される一過性の側鎖構造である.PIGN, PIGO, PIGGはエタノールアミンリン酸転移酵素ファミリーを形成しており,PIGFは,PIGOとPIGGに結合して安定化させる.哺乳動物細胞におけるGPIアンカー前駆体の構造は,EtNP-6Manα1,2(EtNP-6)Manα1,6(EtNP-2)Manα1,4GlcNα1,6(acyl-2)myoInositol1phospho-3(alkyl/acyl-1, acyl-2)glycerolである.
タンパク質C末端にあるGPI付加シグナルペプチドと末端にエタノールアミンを持つGPIアンカー前駆体の入れ替えはアミド基転移反応であり,GPIトランスアミダーゼによって触媒される.GPIトランスアミダーゼは,PIGK, GPAA1, PIGS, PIGT, PIGUの計5成分で複合体酵素を作っている(図1)4).GPIが付加されるアミノ酸をω位(ω-site)アミノ酸と呼ぶ.PIGKはシステインプロテアーゼで,ω位アミノ酸とω+1位アミノ酸間のペプチド結合を切断し,ω位アミノ酸とチオエステル結合した基質酵素中間体を作る.GPAA1は,GPIアンカー前駆体末端のアミノ基とチオエステル中間体の反応を触媒し,アンカーをω位アミノ酸とアミド結合させる.PIGTは,PIGKとジスルフィド結合している.PIGS, PIGUの機能は不明であるが,5成分はいずれもGPI付加に必須である.
できたGPIアンカー型タンパク質はさらに小胞体とゴルジ体において脂質部分と糖鎖部分のリモデリングを受け,成熟型のアンカーを持つGPIアンカー型タンパク質となって,細胞表面に発現する4).
GPIの生合成が全身で欠損すると初期発生が異常になり,胎生致死である9).PNHでは,成体ができた後,造血幹細胞で欠損が起こり,血液系だけに異常細胞が出現するため,溶血性疾患として現れる.
6. PNHの異常血球ではPIGAの体細胞突然変異によってGPIアンカーが欠損する
PNHの異常細胞では,GPI生合成の第1ステップに異常がある10–12).このステップを触媒するGPI-N-アセチルグルコサミン転移酵素は,七つのタンパク質からできた複合体酵素であることをすでに記したが,PNHの異常細胞では,このうちのPIGA遺伝子に機能喪失型の突然変異が起こっている.PIGAは,細胞質側に糖転移酵素ドメインを持つ1回膜貫通型の小胞体タンパク質で,GPI-N-アセチルグルコサミン転移酵素の触媒サブユニットである.PIGA遺伝子の突然変異によって糖転移酵素活性が消失あるいは大きく低下するため,GPI生合成が完全欠損あるいは大きく低下している13).GPI生合成欠損によりアンカー付加が起こらず,C末端の疎水性シグナルペプチドを保持したままになったDAFやCD59などの前駆体タンパク質は,細胞内で,おそらくは小胞体関連分解によって分解される.そのため,すべてのGPIアンカー型タンパク質が細胞表面に発現できない.
PIGA遺伝子の突然変異は体細胞変異である13).PIGA遺伝子は,X染色体上に存在する13).そのため,男性の細胞では一つしか遺伝子が存在しないので,一つの体細胞突然変異によってGPI欠損細胞ができる(図2).女性であっても,X染色体不活性化によって一方のアレルが不活性化されるため,活性型アレルにヒットすればやはり一つの体細胞突然変異でGPI欠損となる(図2).実際,女性患者由来のPNH型Bリンパ芽球では,片方のアレルだけに変異があり,変異を持つアレル由来のmRNAだけが検出された13).
このことは,PNH患者の男女比がほぼ1対1であることと一致している(タイと中国では男性患者が2~3倍多いとの報告があるが14, 15),医療へのアクセスあるいは異なる生活習慣など社会的な要因を反映しているのではなかろうか).
PIGA遺伝子の体細胞変異は,同じ変異がPNH型のBリンパ芽球と好中球に存在すること13),また同じ変異が10年後にも証明されたことから16),長期持続性の多能性造血幹細胞に起こっていることがあらためて確認された.
100例以上のPNH患者のPIGA変異をまとめたデータでは,遺伝子全体や大きな部分を失う変異はまれであって,多くが小さな変異である.変異は患者ごとにさまざまであり,遺伝子内に変異が集中する明確な部位はなく,タンパク質をコードするエクソンとスプライシング部位に広く分布している17).約3分の1ずつが1塩基欠失と1塩基置換,残りの3分の1が1塩基挿入,数塩基の欠失か挿入あるいは,1から数塩基の欠失と挿入の組合わせである.これら体細胞変異の効果は多くがフレームシフトで,その他はスプライシング異常,ナンセンス変異,アミノ酸置換である17).
7. PIGA遺伝子の体細胞変異はPNHの異常細胞に特有な現象ではない
多くの健常者の末梢血好中球には,0.002%程度のCD59欠損細胞が存在し,これらの好中球にはPNHと同様なPIGAの体細胞変異が証明できる.体細胞変異は,数か月後に同じものが証明できることからある程度長期間存在する前駆細胞で起こっていることが報告されている18, 19).また過半数の特発性再生不良性貧血患者(AA)の末梢血好中球ではPNH型細胞の割合が0.003%以上あり20),PIGAの体細胞変異が頻繁にみられる21).これらのことから,PNH細胞でみられるPIGAの体細胞変異の多くは,おそらくは生理的な造血環境下で起こっている遺伝子変異を示していると思われ,PIGA遺伝子変異が直ちにGPIアンカー型タンパク質欠損という表現型で現れ,フローサイトメトリーで鋭敏に検出できることから,観察されると考えられる(図2)19).
PNH患者では,大きく拡大したPIGA変異クローン以外に,別のPIGA体細胞変異を持つクローンが一つないし三つ併存している例が少なくとも20%程度はあることがわかっていた22, 23).最近の牧島らのエクソーム解析の結果では複数クローンを持つ例の比率はもっと高く,二つのクローンが大きく拡大している例もまれではないことが示された24).
これまでに解析されたほとんどのPNH症例においてPIGAの体細胞変異だけが証明され,その他のGPI生合成遺伝子の変異が原因のPNH症例は長く報告されなかった.それは,PIGAだけがX染色体遺伝子で,その他のすべてが常染色体遺伝子であることで説明される.常染色体遺伝子は両アレルが機能しているので,GPI欠損幹細胞になるには二つの体細胞変異が一つの細胞に起こる必要があるが,そのようなことが起こる確率は非常に低いためである.
従来から,もしPIGA以外のGPI生合成遺伝子の変異でPNHが起こるとすれば,一方のアレルに胚細胞性変異があり,造血幹細胞において同じ遺伝子のもう一方のアレルに体細胞変異が起こる場合であると考えられてきた.実際にそのような例が報告された25).この症例は,20番染色体に存在するPIGT遺伝子にスプライシング異常を起こす胚細胞変異があり,PNH型細胞においてもう一方のアレルのPIGT遺伝子全体が欠失していた.つまり,胚細胞変異と体細胞変異の組合わせで,GPIアンカー欠損が起こった1例である.
PIGTは,GPIトランスアミダーゼの構成成分である(図1)26).PIGT欠損細胞ではGPIトランスアミダーゼが失活し,GPIが生合成されているにも関わらず,タンパク質に付加されないため,前駆体タンパク質は分解され,細胞表面へ発現されないので,PNH型細胞になる27).PIGA変異細胞とPIGT変異細胞は,細胞表面でGPIアンカー型タンパク質が欠損していることは共通であるが,生化学的な異常には大きな違いがある.PIGAはGPI生合成の第1ステップに働くため,PIGA変異細胞では,ホスファチジルイノシトールがGPI生合成に使われないだけの状態である.GPIに用いられるのは細胞のホスファチジルイノシトールのごく一部であるため,そのことが細胞の生理に大きな影響を与えることはないと考えられる.一方,PIGTは,GPIをタンパク質に付加する段階に働くため,PIGT変異細胞では,生合成されたGPIがタンパク質のアンカーとして使われずに蓄積する.PIGT変異によるPNH患者は,血管内溶血に加え,じんましん,関節痛などの炎症症状を伴っていることが特徴的である25).GPIの異常蓄積と炎症症状の間にどのような関係があるか今後の研究が必要である.
PNHクローンが拡大して発症に至るメカニズムに関して大きく二つが提唱され,それらを支持するさまざまな結果が報告されたが,現状ではまだ完全理解には至っていない.一つのメカニズムは,PNHがAAと合併するなど,骨髄不全が主要な症状の一つであることに基づいた「骨髄不全状態でPNHクローンが選択的に残って拡大する」という選択説である28, 29).AAが造血幹細胞に対する自己免疫機序で起こることから,GPIアンカー型タンパク質を欠損した細胞が正常細胞より自己免疫に耐性があり,結果生き残るというメカニズムである(図2).上述したように,PIGAの体細胞変異は正常骨髄環境でも生じていて,選択が働くなど第二の事象がなければ拡大は起こらない18, 19).Pigaノックアウトマウスの実験からもこれは裏づけられている30).PNHの骨髄不全そしてAAにおける自己抗原の実体と,抗原認識と細胞障害に関与するリンパ球に関しては不明確な状態であり,メカニズムを決定的に示した研究が待たれる.近年Luzzattoらは,多くのPNH患者に特定のT細胞受容体を持つNKT様のCD4陽性リンパ球クローンが存在し,それが正常幹細胞のCD1d上に提示されたGPIアンカーを認識し障害するというメカニズムを報告している31).PIGA変異細胞はGPIを提示しないので障害されずに選択される32).
もう一つのメカニズムは,PIGA変異幹細胞自体にさらに遺伝子変異が重なって良性腫瘍性を獲得し拡大するとする説である33).AAから発症したPNH症例では,PNHクローンの割合が20から30%程度で長く推移することが多い.一方,多くの症例でPNHクローンが90%以上を占めている.そのとき,一つのクローンが優勢で,小さなクローンが同時に存在することも多い.これら大きさの違うクローンにおいてPIGA変異の種類に違いは認められないので,クローン自体にPIGA変異以外の要因が加わり,その増殖性に変化を生じていると思われる(図2).
井上らは,PNHクローンに12番染色体異常が重なった特異なPNH2例を解析し,良性腫瘍の原因遺伝子として知られているHMGA2遺伝子の変異を見いだした34).2例ともにおいて,HMGA2の片方のアレルが成人でのlet7マイクロRNAを介した発現抑制に必須な3′非翻訳領域を失っており,骨髄において異所性に発現していたことから,PIGA変異幹細胞が良性腫瘍性を獲得したと考えられた34).一方,βサラセミアの遺伝子治療例から,治療用レンチウイルスベクターのイントロンへの挿入によりHMGA2が活性化し,そのクローンが10%程度を占めるまで拡大した例が報告され,PNHの2例での現象と同様の機序が考えられた35).大きく拡大したクローンを持つPNH症例の60%程度で好中球におけるHMGA2 mRNAレベルの有意な上昇がみられ,増殖性の進展をうかがわせる36).しかし,エクソーム解析からは,HMGA2遺伝子に体細胞変異はみられなかった24)ので,制御異常を起こす別の事象が存在すると思われる.
この多数のPNH症例のエクソーム解析から,クローンあたりPIGAの体細胞変異以外に平均二つのさまざまな体細胞変異が存在することがわかった24).その中には,増殖性と関わることが知られている遺伝子に変異がある例があったが,多くの症例の拡大を説明しうる有力な遺伝子は今のところ見いだされていない.エクソーム解析では検出できない遺伝子変異,あるいはエピジェネティックな変異の可能性も残る.
選択説と良性腫瘍説は相互に排他的ではない.AAからPNHクローンが一定の拡大をし,溶血症状を呈するに至るには,選択メカニズムだけで十分かもしれない.しかし,クローンが大きく拡大した典型的なPNHにおいては,増殖性の獲得が起こっていると思われる.骨髄不全の環境下で拡大したPIGA変異幹細胞は,増殖を強いられ,やがて腫瘍性を与える第二の変異を起こし,良性腫瘍性を獲得したサブクローンが大きく拡大するのではないだろうか(図2).
10. 抗補体薬によるPNHの治療とそこからみえてきた新知見
PNHの血管内溶血を抑えるため,ヒト化抗C5モノクローナル抗体医薬であるエクリズマブが用いられている.エクリズマブが結合したC5は,C5転換酵素によるC5aとC5bへの切断が起こらない.そのため,血中に十分な濃度のエクリズマブが維持されると,MAC形成が起こらないため血管内溶血がほぼ完全に抑制される.その結果,輸血必要性の軽減,遊離ヘモグロビンによる諸症状の低減などQOLの大きな改善がみられる37–39).
エクリズマブによる治療が行われることで,従来認識されていなかった2点が明確になった.一つは,エクリズマブが結合できない変異C5を産生する遺伝子多型である.エクリズマブ治療を受けた日本人PNH患者の3%において,血管内溶血がまったく阻害されなかった.これらの人のC5遺伝子の片方のアレルには,885番のアルギニンをヒスチジンに変化させる1塩基多型が共通に存在した40).885番のアルギニンはエクリズマブが認識するエピトープの近傍にあり,変異C5には,エクリズマブはまったく結合しない41–43).変異C5は正常な溶血活性を持つので,これらの人のC5の半分程度にはエクリズマブが結合できず,エクリズマブが十分に存在しても溶血が阻害されない.この遺伝子多型は,日本人健常者にもPNH患者と同じく3%のヘテロ接合体が見つかり,人種的特徴であることが示された40).
二つ目は,血管外溶血である.エクリズマブにより血管内溶血が阻害されているにも関わらず,貧血の改善が十分でなく,骨髄不全では説明がつかないケースがあることから,脾臓での血管外溶血が疑われた.実際,赤血球上に結合したC3断片をフローサイトメーターで測定すると,エクリズマブ治療中の多くの患者のPNH赤血球にC3断片が検出された44).患者の正常赤血球には検出されず,エクリズマブ治療を受けていない患者ではPNH血球にも検出されなかった44).PNH赤血球はDAFが欠損していることにより,いったんC3転換酵素ができると不活化までに時間を要し,その間に産生されたC3bが血球上に次々と結合する.C3転換酵素にC3bが加わることにより,反応特異性がC5転換酵素へと変化する.エクリズマブが存在しないと,C5転換酵素によってC5bが生成され,MAC形成へと進み速やかに溶血する.しかし,ここにエクリズマブが存在すると,C5bが生成されないため,PNH赤血球は溶血を免れ,C5転換酵素を保持し,さらに新たにC3転換酵素ができ得る状態で長期間血管内に存在することになる.そのため,血球上にC3bの断片が蓄積していく.実際,フローサイトメーターで測定するとエクリズマブ開始から日が経つにつれてC3断片の量が増える44).一部の症例では,直接クームス試験でも陽性になる45).脾臓のマクロファージは,補体受容体であるCR3(CD11b/CD18)とCR1(CD35)を発現しており,これらがC3断片に結合してPNH赤血球が捕捉され,そして貪食される46).このようなメカニズムで起こる血管外溶血のため,おそらくは貧血の改善が十分にみられない症例がでると考えられている.
引用文献References
1) Parker, C., Omine, M., Richards, S., Nishimura, J., Bessler, M., Ware, R., Hillmen, P., Luzzatto, L., Young, N., Kinoshita, T., Rosse, W., Socie, G., & International, P.N.H.I.G. (2005) Blood, 106, 3699–3709.
2) Brodsky, R.A. (2014) Blood, 124, 2804–2811.
3) Ware, R.E., Hall, S.E., & Rosse, W.F. (1991) N. Engl. J. Med., 325, 991–996.
4) Kinoshita, T. & Fujita, M. (2016) J. Lipid Res., 57, 6–24.
5) Masuishi, Y., Kimura, Y., Arakawa, N., & Hirano, H. (2016) J. Proteomics, 139, 77–83.
6) Kinoshita, T. (2014) Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 90, 130–143.
7) Miyata, T., Takeda, J., Iida, Y., Yamada, N., Inoue, N., Takahashi, M., Maeda, K., Kitani, T., & Kinoshita, T. (1993) Science, 259, 1318–1320.
8) Watanabe, R., Murakami, Y., Marmor, M.D., Inoue, N., Maeda, Y., Hino, J., Kangawa, K., Julius, M., & Kinoshita, T. (2000) EMBO J., 19, 4402–4411.
9) Nozaki, M., Ohishi, K., Yamada, N., Kinoshita, T., Nagy, A., & Takeda, J. (1999) Lab. Invest., 79, 293–299.
10) Takahashi, M., Takeda, J., Hirose, S., Hyman, R., Inoue, N., Miyata, T., Ueda, E., Kitani, T., Medof, M.E., & Kinoshita, T. (1993) J. Exp. Med., 177, 517–521.
11) Hillmen, P., Bessler, M., Mason, P.J., Watkins, W.M., & Luzzatto, L. (1993) Proc. Natl. Acad. Sci. USA, 90, 5272–5276.
12) Hidaka, M., Nagakura, S., Horikawa, K., Kawaguchi, T., Iwamoto, N., Kagimoto, T., Takatsuki, K., & Nakakuma, H. (1993) Biochem. Biophys. Res. Commun., 191, 571–579.
13) Takeda, J., Miyata, T., Kawagoe, K., Iida, Y., Endo, Y., Fujita, T., Takahashi, M., Kitani, T., & Kinoshita, T. (1993) Cell, 73, 703–711.
14) Kruatrachue, M., Wasi, P., & Na-Nakorn, S. (1978) Br. J. Haematol., 39, 267–276.
15) 張之南(1991)中華内科雑誌,30, 276.
16) Nishimura Ji, J., Hirota, T., Kanakura, Y., Machii, T., Kageyama, T., Doi, S., Wada, H., Masaoka, T., Kanayama, Y., Fujii, H., Inoue, N., Kuwayama, M., Ohishi, K., & Kinoshita, T. (2002) Blood, 99, 2748–2751.
17) Nafa, K., Bessler, M., Castro-Malaspina, H., Jhanwar, S., & Luzzatto, L. (1998) Blood Cells Mol. Dis., 24, 370–384.
18) Araten, D.J., Nafa, K., Pakdeesuwan, K., & Luzzatto, L. (1999) Proc. Natl. Acad. Sci. USA, 96, 5209–5214.
19) Rondelli, T., Berardi, M., Peruzzi, B., Boni, L., Caporale, R., Dolara, P., Notaro, R., & Luzzatto, L. (2013) PLoS One, 8, e54046.
20) Wang, H., Chuhjo, T., Yamazaki, H., Shiobara, S., Teramura, M., Mizoguchi, H., & Nakao, S. (2001) Eur. J. Haematol., 66, 200–205.
21) Yoshizato, T., Dumitriu, B., Hosokawa, K., Makishima, H., Yoshida, K., Townsley, D., Sato-Otsubo, A., Sato, Y., Liu, D., Suzuki, H., Wu, C.O., Shiraishi, Y., Clemente, M.J., Kataoka, K., Shiozawa, Y., Okuno, Y., Chiba, K., Tanaka, H., Nagata, Y., Katagiri, T., Kon, A., Sanada, M., Scheinberg, P., Miyano, S., Maciejewski, J.P., Nakao, S., Young, N.S., & Ogawa, S. (2015) N. Engl. J. Med., 373, 35–47.
22) Endo, M., Ware, R., Vreeke, T., Singh, S., Howard, T., Tomita, A., Holguin, M., & Parker, C. (1996) Blood, 87, 2546–2557.
23) Nishimura, J., Inoue, N., Wada, H., Ueda, E., Pramoonjago, P., Hirota, T., Machii, T., Kageyama, T., Kanamaru, A., Takeda, J., Kinoshita, T., & Kitani, T. (1997) Blood, 89, 3470–3476.
24) Shen, W., Clemente, M.J., Hosono, N., Yoshida, K., Przychodzen, B., Yoshizato, T., Shiraishi, Y., Miyano, S., Ogawa, S., Maciejewski, J.P., & Makishima, H. (2014) J. Clin. Invest., 124, 4529–4538.
25) Krawitz, P.M., Hochsmann, B., Murakami, Y., Teubner, B., Kruger, U., Klopocki, E., Neitzel, H., Hoellein, A., Schneider, C., Parkhomchuk, D., Hecht, J., Robinson, P.N., Mundlos, S., Kinoshita, T., & Schrezenmeier, H. (2013) Blood, 122, 1312–1315.
26) Ohishi, K., Inoue, N., & Kinoshita, T. (2001) EMBO J., 20, 4088–4098.
27) Murakami, Y., Kanzawa, N., Saito, K., Krawitz, P.M., Mundlos, S., Robinson, P.N., Karadimitris, A., Maeda, Y., & Kinoshita, T. (2012) J. Biol. Chem., 287, 6318–6325.
28) Rotoli, B. & Luzzatto, L. (1989) Baillieres Clin. Haematol., 2, 113–138.
29) Young, N.S. (1992) Blood, 79, 1385–1392.
30) Murakami, Y., Kinoshita, T., Maeda, Y., Nakano, T., Kosaka, H., & Takeda, J. (1999) Blood, 94, 2963–2970.
31) Gargiulo, L., Papaioannou, M., Sica, M., Talini, G., Chaidos, A., Richichi, B., Nikolaev, A.V., Nativi, C., Layton, M., de la Fuente, J., Roberts, I., Luzzatto, L., Notaro, R., & Karadimitris, A. (2013) Blood, 121, 2753–2761.
32) Luzzatto, L. (2016) F1000 Res. 10.12688/f1000research.7288.1
33) Kinoshita, T. & Inoue, N. (2002) Int. J. Hematol., 75, 117–122.
34) Inoue, N., Izui-Sarumaru, T., Murakami, Y., Endo, Y., Nishimura, J., Kurokawa, K., Kuwayama, M., Shime, H., Machii, T., Kanakura, Y., Meyers, G., Wittwer, C., Chen, Z., Babcock, W., Frei-Lahr, D., Parker, C.J., & Kinoshita, T. (2006) Blood, 108, 4232–4236.
35) Cavazzana-Calvo, M., Payen, E., Negre, O., Wang, G., Hehir, K., Fusil, F., Down, J., Denaro, M., Brady, T., Westerman, K., Cavallesco, R., Gillet-Legrand, B., Caccavelli, L., Sgarra, R., Maouche-Chretien, L., Bernaudin, F., Girot, R., Dorazio, R., Mulder, G.J., Polack, A., Bank, A., Soulier, J., Larghero, J., Kabbara, N., Dalle, B., Gourmel, B., Socie, G., Chretien, S., Cartier, N., Aubourg, P., Fischer, A., Cornetta, K., Galacteros, F., Beuzard, Y., Gluckman, E., Bushman, F., Hacein-Bey-Abina, S., & Leboulch, P. (2010) Nature, 467, 318–322.
36) Murakami, Y., Inoue, N., Shichishima, T., Ohta, R., Noji, H., Maeda, Y., Nishimura, J., Kanakura, Y., & Kinoshita, T. (2012) Br. J. Haematol., 156, 383–387.
37) Hillmen, P., Young, N.S., Schubert, J., Brodsky, R.A., Socie, G., Muus, P., Roth, A., Szer, J., Elebute, M.O., Nakamura, R., Browne, P., Risitano, A.M., Hill, A., Schrezenmeier, H., Fu, C.L., Maciejewski, J., Rollins, S.A., Mojcik, C.F., Rother, R.P., & Luzzatto, L. (2006) N. Engl. J. Med., 355, 1233–1243.
38) Hillmen, P., Muus, P., Roth, A., Elebute, M.O., Risitano, A.M., Schrezenmeier, H., Szer, J., Browne, P., Maciejewski, J.P., Schubert, J., Urbano-Ispizua, A., de Castro, C., Socie, G., & Brodsky, R.A. (2013) Br. J. Haematol., 162, 62–73.
39) Kanakura, Y., Ohyashiki, K., Shichishima, T., Okamoto, S., Ando, K., Ninomiya, H., Kawaguchi, T., Nakao, S., Nakakuma, H., Nishimura, J., Kinoshita, T., Bedrosian, C.L., Valentine, M.E., Khursigara, G., Ozawa, K., & Omine, M. (2011) Int. J. Hematol., 93, 36–46.
40) Nishimura, J., Yamamoto, M., Hayashi, S., Ohyashiki, K., Ando, K., Brodsky, A.L., Noji, H., Kitamura, K., Eto, T., Takahashi, T., Masuko, M., Matsumoto, T., Wano, Y., Shichishima, T., Shibayama, H., Hase, M., Li, L., Johnson, K., Lazarowski, A., Tamburini, P., Inazawa, J., Kinoshita, T., & Kanakura, Y. (2014) N. Engl. J. Med., 370, 632–639.
41) Jore, M.M., Johnson, S., Sheppard, D., Barber, N.M., Li, Y.I., Nunn, M.A., Elmlund, H., & Lea, S.M. (2016) Nat. Struct. Mol. Biol., 23, 378–386.
42) Schatz-Jakobsen, J.A., Zhang, Y., Johnson, K., Neill, A., Sheridan, D., & Andersen, G.R. (2016) J. Immunol., 197, 337–344.
43) Volk, A.L., Hu, F.J., Berglund, M.M., Nordling, E., Stromberg, P., Uhlen, M., & Rockberg, J. (2016) Sci. Rep., 6, 31365.
44) Risitano, A.M., Notaro, R., Marando, L., Serio, B., Ranaldi, D., Seneca, E., Ricci, P., Alfinito, F., Camera, A., Gianfaldoni, G., Amendola, A., Boschetti, C., Di Bona, E., Fratellanza, G., Barbano, F., Rodeghiero, F., Zanella, A., Iori, A.P., Selleri, C., Luzzatto, L., & Rotoli, B. (2009) Blood, 113, 4094–4100.
45) Subias Hidalgo, M., Martin Merinero, H., Lopez, A., Anter, J., Garcia, S.P., Ataulfo Gonzalez-Fernandez, F., Fores, R., Lopez-Trascasa, M., Villegas, A., Ojeda, E., & Rodriguez de Cordoba, S. (2017) Immunobiology, 222, 363–371.
46) Lin, Z., Schmidt, C.Q., Koutsogiannaki, S., Ricci, P., Risitano, A.M., Lambris, J.D., & Ricklin, D. (2015) Blood, 126, 891–894.
著者紹介Author Profile
 木下 タロウ(きのした たろう)
木下 タロウ(きのした たろう)大阪大学微生物病研究所籔本難病解明寄附研究部門教授.医学博士.
略歴1951年兵庫県に生る.74年東京大学農学部卒業.77年同大学院農学系研究科修士課程修了.81年大阪大学大学院医学研究科博士課程修了.同年日本学術振興会奨励研究員.82年大阪大学医学部助手(細菌学).88年同講師.90年大阪大学微生物病研究所免疫不全疾患研究分野教授.2003~07年同研究所長.07年大阪大学免疫学フロンティア研究センター副拠点長,教授(糖鎖免疫学研究室).17年から現職.
研究テーマタンパク質GPIアンカーの生物学と医学.
抱負糖脂質であるGPIがタンパク質の膜アンカーに用いられることの生理的意義を理解し,合わせてGPI欠損により起こる諸疾患の病態を解明したい.
ウェブサイトhttp://www.biken.osaka-u.ac.jp/biken/men-eki-huzen/index.html