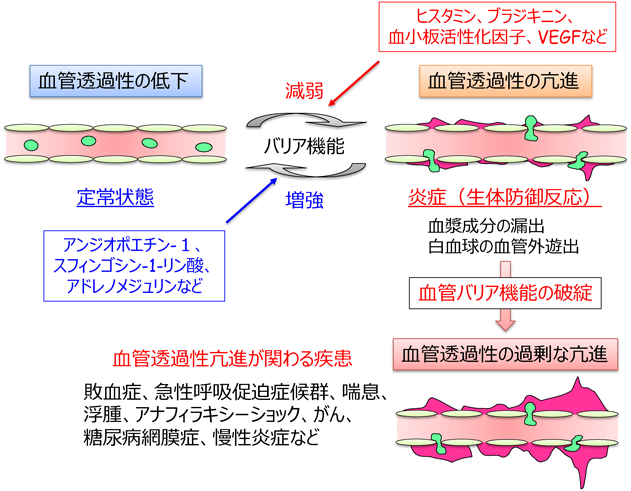
血管の内腔面でシート構造を形成する血管内皮細胞は,血液–組織間の物質移動,すなわち“血管透過性”を厳密に制御することにより,生体の恒常性維持に寄与している(図1).通常,正常組織では血管透過性は低い状態に維持されているが,炎症が誘導されると,生体防御反応として血管透過性が亢進し,免疫細胞の血管外への移動や血漿成分の漏出が惹起される.また,がん細胞は内皮細胞の間隙を通り抜けて,原発巣から他の組織,臓器に浸潤・転移する.そのため内皮細胞には,血管透過性をダイナミックかつ厳密に制御するための仕組みが備わっており,その制御機構の破綻は,敗血症,急性呼吸促迫症候群,喘息,浮腫,アナフィラキシーショック,がん,糖尿病網膜症,慢性炎症などさまざまな疾患と密接に関連している1)(図1).
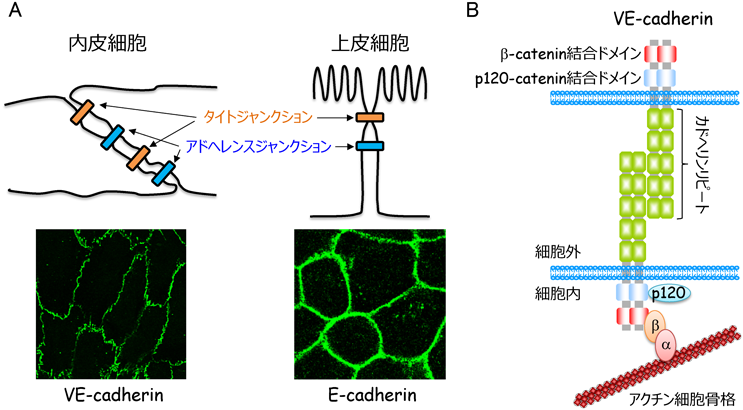
血管壁を介した物質移動には,細胞内を通り抜けるトランスセルラー経路と細胞間隙を通るパラセルラー経路が存在する2).前者は,主にカベオラ依存性エンドサイトーシスに依存しており,後者は内皮細胞間接着によって制御されている.内皮細胞間接着は,アドヘレンスジャンクション(AJ)とタイトジャンクション(TJ)の二つの接着装置により形成される3).内皮細胞と同様にシート構造を形成する上皮細胞は,頂端–基底軸に沿って極性を形成し,細胞接着面の頂端側からTJ, AJを形成することで,バリア機能の高い細胞シートを構築する(図2).一方,内皮細胞は,血液脳関門では循環血液と脳内の物質輸送を制限するため,バリア機能の高い細胞間接着装置を形成するが,末梢組織のほとんどの血管では明瞭な細胞極性を構築せず,TJ, AJが細胞間接着部位で混在している3, 4)(図2).このような内皮細胞に特有の細胞間接着装置は,血管透過性のダイナミックな制御に重要である.
血管透過性の制御には,AJを構成する主要な接着分子であるvascular endothelial(VE)-cadherinが重要な役割をしている3).実際に,VE-cadherinの中和抗体をマウスに静脈注射すると,血管透過性が亢進し,浮腫を来すことが報告されている5).VE-cadherin接着は,血管透過性を調節するさまざまな因子によって,正および負に制御される(図1).たとえば,炎症が誘導されると,ヒスタミンやブラジキニン,血小板活性化因子(PAF),サイトカインなど,さまざまな液性因子が産生されるが,これら因子は,VE-cadherin接着を弱め,血管透過性を亢進する1, 6, 7).また,血管新生因子である血管内皮増殖因子(VEGF)もVE-cadherin接着を破壊することで血管透過性を亢進する8, 9).一方,アンジオポエチン-1(Ang1)や脂質メディエーターであるスフィンゴシン1-リン酸(S1P)は,VE-cadherin接着を増強することで血管透過性を抑制することが知られている10, 11).また,細胞内セカンドメッセンジャーであるサイクリックAMP(cAMP)の産生は,VE-cadherin接着を強め,血管透過性を低下させる12, 13).実際に,cAMP産生活性を有するGタンパク質共役型受容体(GPCR)アゴニストであるアドレノメジュリン,プロスタサイクリン(PGI2),プロスタグランジンE2(PGE2),β-アドレナリン受容体アゴニストは,炎症性メディエーターによる血管透過性の亢進を抑えることが報告されている14–16).
本稿では,VE-cadherin接着の制御機構に焦点を当て,血管透過性のダイナミックかつ巧妙な制御を可能にするシグナル伝達系について,最近の知見を紹介する.特に,VE-cadherinのリン酸化とその血管透過性制御における役割,Rap1およびRhoファミリー低分子量Gタンパク質によるアクチン細胞骨格の再編性によるVE-cadherin接着調節機構,またAng1による血管透過性制御機構について概説する.さらに,これらVE-cadherin接着を制御するシグナル伝達系が,血管透過性の亢進が関わる疾患の治療ターゲットになりうる可能性について紹介する.
2. VE-cadherinが形成する内皮細胞間接着装置
VE-cadherin(別名Cadherin-5, CD144という)は,クラシカルカドヘリンファミリーに属する1回膜貫通型タンパク質で,五つのカドヘリンリピートからなる細胞外ドメイン,膜貫通ドメイン,細胞内ドメインから構成されている3, 6)(図2).VE-cadherinは,カルシウム依存性にシス二量体を形成し,さらに隣接する細胞のシス二量体と細胞外ドメインを介してトランス結合することで強固な細胞間接着を形成する.また,細胞内ドメインの膜貫通領域に隣接する部位にはp120-cateninが,C末端領域にはβ-cateninが結合し,β-cateninはさらにα-cateninと相互作用する.p120-catenin結合領域は,VE-cadherinのエンドサイトーシスに関わる領域と重複しているため,p120-cateninはVE-cadherinのエンドサイトーシスを抑制することで,VE-cadherin接着を安定化する17).一方,VE-cadherin/β-catenin複合体に結合したα-cateninは,直接的あるいはvinculin, α-actinin, epithelial protein lost in neoplasm(EPLIN)などのリンカータンパク質を介して間接的にアクチン細胞骨格と相互作用することで,VE-cadherin接着装置をアクチン細胞骨格につなぎ止めている18–20).そのため,アクチン細胞骨格は,VE-cadherin接着さらには血管透過性の制御にきわめて重要な役割を果たしている.
3. VE-cadherinのリン酸化による血管透過性制御
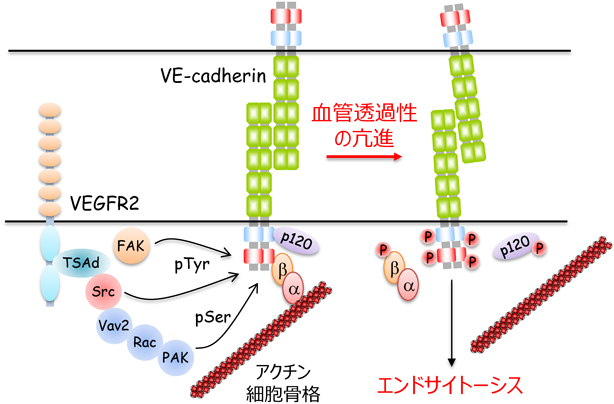
VEGFやヒスタミンなどの炎症性メディエーターは,VE-cadherinの細胞内ドメインやそこに結合するp120-catenin・β-cateninをリン酸化する6, 21).それにより,VE-cadherinとp120-catenin・β-cateninの解離やVE-cadherinのエンドサイトーシスが誘導され,血管透過性が亢進する.また,炎症時に白血球は,血管外に遊出するため,内皮細胞表面のintercellular adhesion molecule-1を介して内皮細胞に接着し,VE-cadherinのリン酸化を惹起する.このように,VE-cadherinのリン酸化は,血管透過性や白血球の血管外遊出の制御に重要な役割を果たしている21).
VEGFは,VEGF受容体2(VEGFR2)を介して,非受容体型チロシンキナーゼSrcを活性化し,血管透過性を亢進する22–24)(図3).VEGFの結合によりVEGFR2は,951番目のチロシン残基を自己リン酸化し,そのリン酸化部位にアダプタータンパク質であるT cell-specific adaptor(TSAd)をリクルートする9, 25, 26).TSAdは,さらにSrcを動員し,活性化することで,VE-cadherinをリン酸化し,血管透過性を亢進する.また,SrcはRhoファミリー低分子量Gタンパク質の一つRacを介してp21-activated kinase(PAK)を活性化し,VE-cadherinのセリン残基をリン酸化する27).PAKによってリン酸化されたVE-cadherinは,β-arrestin依存的にエンドサイトーシスされ,血管透過性が亢進する.加えて,VEGFは別の非受容体型チロシンキナーゼfocal adhesion kinase(FAK)を活性化することで,β-cateninをリン酸化し,血管透過性を亢進する28)(図3).
最近,VE-cadherinの各チロシン残基のリン酸化を特異的に認識する抗体が開発され,VE-cadherin接着制御に関わるチロシン残基の同定が試みられた.Orsenigoらは,定常状態においてVE-cadherinの658・685番目のチロシン残基が静脈でのみリン酸化されていること,また,これらチロシン残基のリン酸化が炎症性メディエーターによる血管透過性の亢進に必要であることを報告した29).Wesselらは,685番目と731番目のチロシン残基をフェニルアラニンに置換したVE-cadherinを発現するノックインマウスを樹立・解析し,血管透過性亢進と白血球の血管外遊出には,VE-cadherinの異なったチロシン残基のリン酸化が重要であることを明らかにした30).白血球は,定常状態でリン酸化されている731番目のチロシン残基を脱リン酸化することで,VE-cadherinのエンドサイトーシスを誘導し,血管外へ遊出する.一方,VEGFやヒスタミンなどの炎症性メディエーターは,VE-cadherinの685番目のチロシン残基をリン酸化することで,血管透過性を亢進する.しかし,Sidibéらは,独自に685番目のチロシン残基をフェニルアラニンに置換したVE-cadherinを発現するノックインマウスを樹立し,このマウスでは恒常的に血管透過性が亢進しており,VEGFによる血管透過性の亢進が増強されていることを示した31).以前のin vitro系の解析から,VE-cadherinの685番目のリン酸化チロシン残基にSrcのインヒビターであるCOOH-terminal Src kinase(Csk)が結合することが示唆されている32).これらの知見は,VE-cadherinの685番目のチロシン残基のリン酸化が,血管透過性の亢進に対して抑制的に働く可能性を示唆しており,今後これら矛盾した結果が得られた原因についてさらなる解析が待たれる.
VE-cadherinのリン酸化レベルは,プロテインキナーゼによるリン酸化に加え,プロテインホスファターゼによる脱リン酸化によっても制御されている.これまで内皮細胞間接着に関わるプロテインホスファターゼとして,VE-protein tyrosine phosphatase(VE-PTP)33, 34),density-enhanced phosphatse-1(DEP-1)35),protein tyrosine phosphatase receptor type M(PTPμ)36)やSrc homology 2-containing protein tyrosine phosphatase(SHP2)37)などが報告されている.特にVE-PTPは,内皮細胞に特異的に発現するプロテインホスファターゼであり,VE-cadherinと相互作用し,脱リン酸化状態に維持することで,VE-cadherin接着を安定化することが知られている33, 34).そのため,VEGFや白血球は,VE-cadherinからVE-PTPを解離することで,VE-cadherinを効率的にリン酸化し,血管透過性の亢進や白血球の血管外移出を惹起する34, 38).
4. 炎症性メディエーターはRhoを介して血管透過性を亢進する
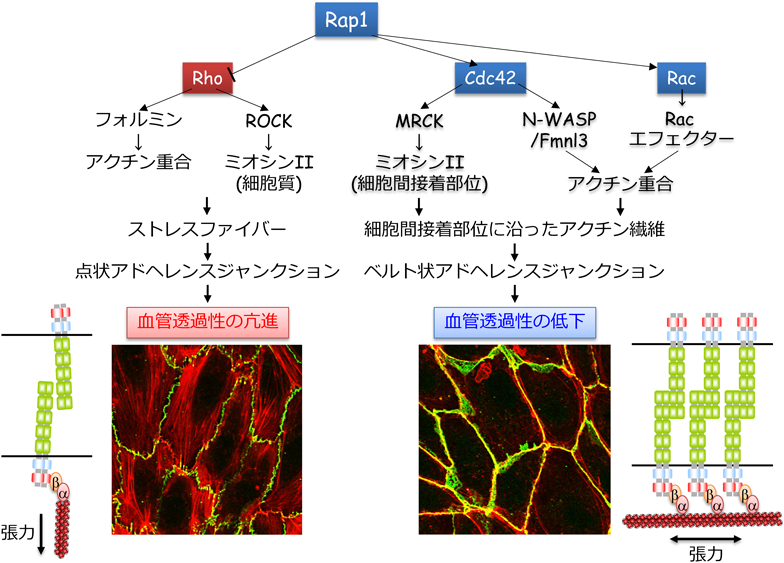
血管透過性亢進作用を有するヒスタミン,ブラジキニン,PAF,トロンビンなどの炎症性メディエーターは,Rho低分子量Gタンパク質を介して,細胞質に収縮性アクチン細胞骨格であるストレスファイバーを形成し,内皮細胞の収縮を惹起する39–42).それに伴い,VE-cadherin接着部位に張力がかかり,VE-cadherinはその張力に抵抗するため,放射状に配置されたアクチン繊維の束(radial actin bundle)に結合し,不連続な点状の接着構造を形成する.この接着構造を点状AJ(punctate AJ)と呼び,その形成は血管透過性の亢進と相関する.
Rhoは,アクチン重合促進活性を有するフォルミンタンパク質を介してアクチン繊維を形成する.また,その一方でRhoは,Rhoキナーゼ(ROCK)を介してミオシンIIを活性化し,フォルミンによって形成されたアクチン繊維に張力を負荷することで,収縮性のアクチン繊維であるストレスファイバーを形成する39, 41, 43, 44)(図4).すなわち,炎症性メディエーターは,Rhoを介してストレスファイバーを形成し,内皮細胞を収縮することで,VE-cadherin接着の減弱と血管透過性の亢進を惹起する(図4).
5. Rap1はVE-cadherin接着を増強し血管透過性を抑制する
昔から内皮細胞におけるcAMPの産生が,血管透過性の低下をもたらすことが知られていた12, 13).以前,我々と他のグループは,その分子メカニズムについて解析を行い,cAMPの下流で血管透過性を制御する因子としてRap1を同定した45–47).Rap1は,Rasファミリーに属する低分子量Gタンパク質で,Rasによる細胞の形質転換を抑制する分子として同定された48).しかし,その後の解析から,Rap1はintegrin活性化能を有し,細胞–基質間接着を制御することが示された49).Rap1をはじめとするGタンパク質は,グアニンヌクレオチド交換因子(GEF)によってGDP結合型からGTP結合型に変換されることで活性化する.cAMPは,protein kinase A(PKA)とRap1のGEFであるexchange protein directly activated by cAMP(Epac)の二つのエフェクターを活性化する.そこで,どちらのエフェクター分子がcAMPによる血管透過性制御に関与するか検討したところ,cAMPはEpacを介してRap1を活性化し,VE-cadherin接着の増強と血管透過性の低下を引き起こすことが示された45–47).また,Rap1はVE-cadherin接着自身によっても活性化され,VE-cadherin接着の成熟化に関与する50, 51).VE-cadherinは,β-cateninを介してアダプタータンパク質であるMAGI-1を細胞間接着部位にリクルートする.MAGI-1は,さらにRap1のGEFの一つであるPDZ-GEFを動員することで細胞間接着部位でRap1を活性化し,VE-cadherin接着を安定化する.さらに,内皮細胞特異的Rap1欠損マウスは浮腫を呈することから,Rap1が生体内でも内皮細胞間接着に関与することが示唆されている52).これらの研究から,Rap1はintegrinを介した細胞–基質間接着とcadherinを介した細胞間接着の両方を制御するシグナル分子であることが明らかになった.実際に,Rap1が上皮細胞におけるE-cadherin接着にも関与することが報告されている53–55).
6. Rap1はアクトミオシン活性を空間的に制御することでVE-cadherin接着を増強する
Rap1は,点状AJを破壊し,その一方で,細胞間接着部位に沿ったアクチン繊維の束(circumferential actin bundle:CAB)を形成することで,VE-cadherin接着を増強する.CABは,α-/β-cateninを介してVE-cadherinを細胞間接着部位にアンカーし,バリア機能の高い細胞間接着装置を構築する19, 46).このような接着構造をベルト状AJ(linear AJ)と呼び,その形成は血管透過性の低下と相関する.VE-cadherinとα-cateninの融合タンパク質を発現するノックインマウスでは,白血球の血管外遊出や血管透過性の亢進が抑えられることが報告されており,α-cateninを介したVE-cadherinのアクチン細胞骨格への結合がVE-cadherin接着の安定化に重要であることがin vivo実験でも証明されている18).すなわち,Rap1は点状AJを破壊し,ベルト状AJを形成することで,VE-cadherin接着を強め,血管透過性を抑えると考えられる(図4).
Rap1は,Rhoシグナルを抑制することで,アクチンストレスファイバーの形成を抑え,点状AJを破壊する.脳海綿状血管腫(cerebral cavernous malformation)は,出血を伴う血管奇形の一種で,その原因遺伝子の一つとしてRap1のエフェクター分子であるCCM1/Krev interacting trapped protein-1(Krit-1)が報告されている.CCM1/Krit-1がRhoシグナルを抑制することが報告されており,Rap1によるRhoシグナルの制御にCCM1/Krit-1が関与することが示唆されているが,その分子メカニズムの詳細は不明である56, 57).また,最近,Rap1によるRhoの抑制に関わるエフェクター分子としてRas-interacting protein 1(Rasip-1)が同定された58, 59).Rap1は,Rasip-1に結合することで,その結合パートナーであるArhGAP29を活性化する.ArhGAP29は,Rhoに対するGTPase活性化タンパク質であり,Rhoに結合するGTPをGDPに加水分解することでRhoを不活性化状態に変換する.このため,Rap1はRasip-1/ArhGAP29を介してRhoシグナルを抑え,内皮細胞間接着を増強することが示唆されている.
我々はさらに,Rap1がCABを形成し,ベルト状AJを構築する分子メカニズムについて解析を行った.その結果,Rap1は細胞間接着部位でRhoファミリー低分子量Gタンパク質メンバーの一つCdc42を活性化すること,さらにCdc42がmyotonic dystrophy kinase-related CDC42-binding kinase(MRCK)を介してミオシンIIの調節軽鎖をリン酸化し活性化することで,CABの形成維持に関与することを示した60)(図4).Rap1は,Cdc42を細胞間接着部位へリクルートするとともに,Cdc42のGEFの一つFYVE, RhoGEF and PH domain containing 5(FGD5)を介して細胞間接着部位におけるCdc42の活性化を促進し,効率的に活性化型Cdc42を細胞間接着部位に集積させる60).
Rap1によるCdc42-MRCKシグナル伝達系を介した細胞間接着部位におけるミオシンIIの活性化は,CABに張力を負荷し,その形成維持に寄与する.そのため,Rap1はこれとは別の経路を介して,細胞間接着部位におけるアクチン重合を促進しCAB形成を制御すると考えられる(図4).その候補として,Cdc42の下流エフェクター分子であるneuronal Wiskott-Aldrich syndrome(N-WASP)やFormin-like 3(Fmnl3)が考えられる61–64).Fmnl3については,内皮細胞間の接着部位でアクチン繊維を形成し,管腔形成に寄与することが報告されている63).上皮細胞でも,Fmnl3が細胞間接着部位でアクチン繊維を形成し,E-cadherin接着を安定化することが報告されている62).また,Rap1によるCAB形成に関わる候補分子として,Cdc42と同様にRhoファミリーGタンパク質メンバーに属するRacがあげられる.Racは,S1P, PGE2, PGI2,肝細胞増殖因子など血管透過性を抑える働きのある因子によって活性化される10, 44, 65, 66).実際にRap1による血管透過性の抑制に,Rac依存的なアクチン細胞骨格制御が関与すること66),また,Rap1がTiam1やVav2などのRacのGEFに結合し,Rac活性を空間的に制御するという報告がなされている67).しかし,Racが内皮細胞間接着を弱め血管透過性を亢進することも報告されており,今後,Racが血管透過性に対して相反する機能を発揮する機序についての解析が必要である27, 68).さらに,AF6(別名Afadin)やRap1-interacting adaptor molecule(RIAM)などアクチン細胞骨格制御に関わるRap1エフェクターも同定されており69, 70),Rap1によるCAB形成に関わるシグナル伝達機構については,今後のさらなる解析が必要である.
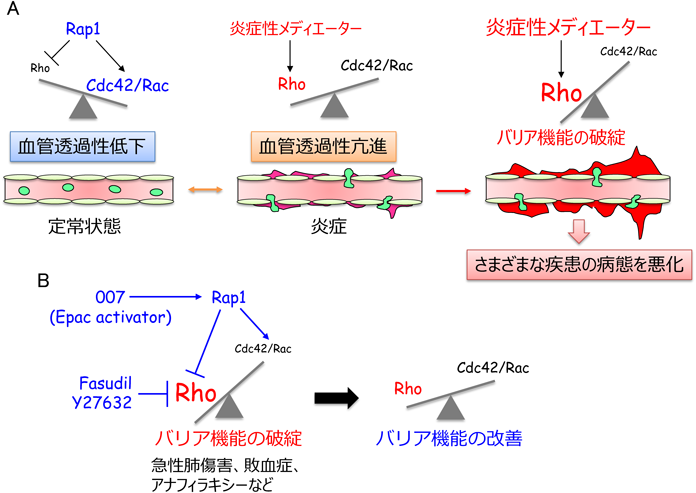
7. Cdc42/RacシグナルとRhoシグナルのバランスが血管透過性を規定しており,その正常化は血管透過性が関わる疾患の病態を緩和する
血管透過性はCdc42/RacシグナルとRhoシグナルのバランスによって規定されており,Rap1はCdc42/Racシグナルを活性化し,その一方でRhoシグナルを抑えることで,血管透過性を抑制する(図5).正常組織では,Rap1活性が比較的高く,それによりCdc42/RacシグナルがVE-cadherin接着を増強し,血管透過性を低い状態に維持していると考えられる.一方,炎症が誘導されると,炎症性メディエーターがRhoを活性化しVE-cadherin接着を減弱することで,血管透過性を亢進する.敗血症,急性呼吸促迫症候群,喘息,浮腫,アナフィラキシーショック,がん,糖尿病網膜症,慢性炎症などの血管透過性の亢進が関わる疾患では,炎症性メディエーターの過剰な産生などが原因でRhoが強力に活性化し,それにより血管透過性が過剰に亢進し,これら疾患の病態を悪化させると考えられる.したがって,RhoシグナルとCdc42/Racシグナルのバランスを制御することができれば,これら血管透過性亢進が関わる疾患の病態を改善できるかもしれない(図5).実際に,モデル実験動物を用いた解析から,この可能性を支持する知見が報告されている.
Epacを特異的に活性化するcAMPアナログ007(8-pCPT-2′-O-Me-cAMP)は,マウスにおいて,VEGFやPAFによる血管透過性の亢進を抑える46, 71).同様に,007によるRap1の活性化が,リポポリサッカライド(LPS)による急性肺傷害や血管透過性亢進を改善する72).また,cAMP産生活性を有するPGI2の誘導体iloprostが,Rap1を介して人工呼吸惹起性肺損傷や血管バリア機能の破綻を緩和することが示されている73).さらに,S1PがRacによるアクチン細胞骨格の再編成を介して,急性肺傷害における血管透過性亢進を抑える74, 75).これら知見は,Rap1の活性化とそれに続くCdc42/Racシグナル依存的なVE-cadherin接着の増強が,血管透過性亢進が関わる疾患の病態を改善する可能性を示唆している.同様に,Rhoシグナル系の阻害が,種々の疾患モデルにおける血管透過性亢進を抑えることが報告されている.Rhoの下流エフェクター分子であるROCKの阻害剤Fasudilが,アナフィラアキシーショックに伴う血管透過性亢進を抑え,アナフィラキシーショックの症状を緩和することが報告された76).また,ROCK阻害剤であるY27632が,LPSによる肺水腫や急性肺傷害を抑えることが示されている77, 78).以上の報告は,RhoシグナルとCdc42/Racシグナルのバランスの正常化が,血管透過性亢進が関わる疾患の治療法となりうることを示唆しており,今後,これらシグナル系をターゲットにした治療薬の開発が期待される.
8. Ang1-Tie2シグナルによる血管透過性制御
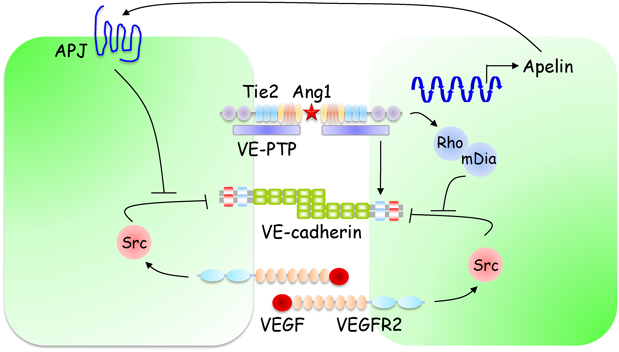
Ang1は,Tie2受容体を介して,内皮細胞間および内皮細胞−壁細胞間接着を増強し,血管安定化と血管透過性の低下を引き起こす79)(図6).一方,Ang1のアンタゴニストであるAng2は,Ang1-Tie2シグナルを遮断し,血管構造の不安定化と血管透過性の亢進を惹起する.Ang1はin vitroの内皮細胞において,VE-cadherin接着を増強し,血管透過性を低下させることが報告されている11).また,Ang1をケラチノサイトで過剰発現するマウスでは,VEGFや炎症性メディエーターによる血管透過性亢進が顕著に抑制されていることから,Ang1-Tie2シグナルはVE-cadherin接着を増強し,血管バリア機能を制御していると考えられる80).
Ang1は,VEGFによるSrcの活性化を阻害することで,VEGFによるVE-cadherin接着破壊および血管透過性亢進を抑制する.Gavardらは,Ang1-Tie2シグナルが,Rho−mDia経路を介して,VEGFによるSrcの活性化を阻害することを報告した81).しかし,RhoがVE-cadherin接着を弱め,血管透過性を亢進すること,また,Ang1による血管透過性抑制に,p190RhoGAPを介したRhoの抑制が重要であるという知見も報告されており82),今後Tie2シグナルにおけるRhoの役割についてさらなる検討が必要である.また,Ang1-Tie2シグナルが,Apelinの発現を介して,VE-cadherin接着の増強と血管透過性の低下を引き起こすことが報告されている83).
Ang1-Tie2シグナルは,血管安定化と血管新生の両方を制御する.以前,我々とAlitaloらのグループは,Ang1-Tie2シグナルがこれら相反する機能を制御する機構について解析を行い,Ang1-Tie2シグナルは,内皮細胞間接着によって空間的・機能的に制御されていることを示した84, 85).細胞間接着非存在下では,Ang1は細胞外基質に結合し,細胞–基質接着面にTie2をアンカーすることで,Erk1/2などの血管新生シグナルを活性化する.一方,細胞間接着を有する内皮細胞では,Ang1は細胞間接着部位でTie2のトランス結合を形成し,Aktなどの血管安定化シグナルを活性化する.また,トランス結合したTie2が,細胞間接着部位でVE-PTPと相互作用することが示唆されている.VE-PTPはVE-cadherinとも相互作用することから,トランス結合したTie2がVE-PTPを介してVE-cadherin接着を増強すると考えられる.
Ang1-Tie2シグナルの活性化が,血管透過性亢進が関わる疾患の治療法となる可能性が示唆されている.LPSによる急性肺傷害モデルにおいて,強力なTie2活性化能を有するAng1誘導体COMP-Ang1が,血管透過性を抑え,肺傷害を軽減することが報告されている86).同様に,Ang1が,脈絡膜における血管新生と血管透過性の亢進を抑えること,また,糖尿病網膜症モデルでは,Ang1が網膜の血管透過性を抑制することが報告されている87, 88).また,最近,Kohらのグループは,Ang2に結合し多量体化することで,Ang2をTie2のアンタゴニストからアゴニストへと変換する抗体(Ang2-binding and Tie2-activating antibody:ABTAA)を開発し,この抗体が敗血症の症状を緩和することを示した89).今後,Ang1-Tie2シグナルをターゲットにした,血管透過性亢進が関わる疾患の治療薬の開発が期待される.
血管内皮細胞は,血管透過性をダイナミックかつ厳密に制御することで生体恒常性を維持しており,その破綻は,さまざまな疾患の発症・進展と密接に関連する.これまで内皮細胞が血管透過性を制御する機構について,数多くの基礎研究がなされ,血管透過性制御に関わるシグナル伝達系が徐々に明らかになりつつある.さらに,これらシグナル伝達機構は,血管透過性亢進が関わる疾患の治療ターゲットになりうる可能性が示唆されている.しかし,今後明らかにしなければならない課題も数多く残されているのも事実である.たとえば,Cdc42/RacシグナルとRhoシグナルのバランスが血管透過性を規定している可能性を述べたが,これら知見は主に内皮細胞を用いたin vitro実験により明らかにされたものであり,実際にこれら機構が生体でも機能していることを検証する必要がある.そのためには,生きた個体における血管透過性を解析することが重要であり,そのためには生体内の細胞機能や分子活性を可視化する蛍光生体イメージング技術が有効である.また,生体内の各臓器では,それぞれの機能に適した血管が構築され,血管透過性の制御機構も臓器によって異なる.そのため,生体恒常性維持機構の解明には,血管透過性の制御機構の多様性を理解することが重要である.今後,これら残された課題が基礎研究によって明らかにされ,その成果が血管透過性亢進が関わる疾患の治療法の開発につながることが期待される.
引用文献References
1) Park-Windhol, C. & D’Amore, P.A. (2016) Annu. Rev. Pathol., 11, 251–281.
2) Komarova, Y. & Malik, A.B. (2010) Annu. Rev. Physiol., 72, 463–493.
3) Dejana, E. (2004) Nat. Rev. Mol. Cell Biol., 5, 261–270.
4) Obermeier, B., Daneman, R., & Ransohoff, R.M. (2013) Nat. Med., 19, 1584–1596.
5) Corada, M., Mariotti, M., Thurston, G., Smith, K., Kunkel, R., Brockhaus, M., Lampugnani, M.G., Martin-Padura, I., Stoppacciaro, A., Ruco, L., McDonald, D.M., Ward, P.A., & Dejana, E. (1999) Proc. Natl. Acad. Sci. USA, 96, 9815–9820.
6) Dejana, E., Orsenigo, F., & Lampugnani, M.G. (2008) J. Cell Sci., 121, 2115–2122.
7) Andriopoulou, P., Navarro, P., Zanetti, A., Lampugnani, M.G., & Dejana, E. (1999) Arterioscler. Thromb. Vasc. Biol., 19, 2286–2297.
8) Weis, S.M. & Cheresh, D.A. (2005) Nature, 437, 497–504.
9) Claesson-Welsh, L. (2015) Ups. J. Med. Sci., 120, 135–143.
10) Garcia, J.G., Liu, F., Verin, A.D., Birukova, A., Dechert, M.A., Gerthoffer, W.T., Bamberg, J.R., & English, D. (2001) J. Clin. Invest., 108, 689–701.
11) Gamble, J.R., Drew, J., Trezise, L., Underwood, A., Parsons, M., Kasminkas, L., Rudge, J., Yancopoulos, G., & Vadas, M.A. (2000) Circ. Res., 87, 603–607.
12) Stelzner, T.J., Weil, J.V., & O’Brien, R.F. (1989) J. Cell. Physiol., 139, 157–166.
13) Moy, A.B., Bodmer, J.E., Blackwell, K., Shasby, S., & Shasby, D.M. (1998) Am. J. Physiol., 274, L1024–L1029.
14) Langeler, E.G. & van Hinsbergh, V.W. (1991) Am. J. Physiol., 260, C1052–C1059.
15) Hippenstiel, S., Witzenrath, M., Schmeck, B., Hocke, A., Krisp, M., Krull, M., Seybold, J., Seeger, W., Rascher, W., Schutte, H., & Suttorp, N. (2002) Circ. Res., 91, 618–625.
16) Farmer, P.J., Bernier, S.G., Lepage, A., Guillemette, G., Regoli, D., & Sirois, P. (2001) Am. J. Physiol. Lung Cell. Mol. Physiol., 280, L732–L738.
17) Xiao, K., Allison, D.F., Buckley, K.M., Kottke, M.D., Vincent, P.A., Faundez, V., & Kowalczyk, A.P. (2003) J. Cell Biol., 163, 535–545.
18) Schulte, D., Kuppers, V., Dartsch, N., Broermann, A., Li, H., Zarbock, A., Kamenyeva, O., Kiefer, F., Khandoga, A., Massberg, S., & Vestweber, D. (2011) EMBO J., 30, 4157–4170.
19) Noda, K., Zhang, J., Fukuhara, S., Kunimoto, S., Yoshimura, M., & Mochizuki, N. (2010) Mol. Biol. Cell, 21, 584–596.
20) Chervin-Petinot, A., Courcon, M., Almagro, S., Nicolas, A., Grichine, A., Grunwald, D., Prandini, M.H., Huber, P., & Gulino-Debrac, D. (2012) J. Biol. Chem., 287, 7556–7572.
21) Vestweber, D., Wessel, F., & Nottebaum, A.F. (2014) Semin. Immunopathol., 36, 177–192.
22) Weis, S., Cui, J., Barnes, L., & Cheresh, D. (2004) J. Cell Biol., 167, 223–229.
23) Eliceiri, B.P., Paul, R., Schwartzberg, P.L., Hood, J.D., Leng, J., & Cheresh, D.A. (1999) Mol. Cell, 4, 915–924.
24) Koch, S., Tugues, S., Li, X., Gualandi, L., & Claesson-Welsh, L. (2011) Biochem. J., 437, 169–183.
25) Sun, Z., Li, X., Massena, S., Kutschera, S., Padhan, N., Gualandi, L., Sundvold-Gjerstad, V., Gustafsson, K., Choy, W.W., Zang, G., Quach, M., Jansson, L., Phillipson, M., Abid, M.R., Spurkland, A., & Claesson-Welsh, L. (2012) J. Exp. Med., 209, 1363–1377.
26) Matsumoto, T., Bohman, S., Dixelius, J., Berge, T., Dimberg, A., Magnusson, P., Wang, L., Wikner, C., Qi, J.H., Wernstedt, C., Wu, J., Bruheim, S., Mugishima, H., Mukhopadhyay, D., Spurkland, A., & Claesson-Welsh, L. (2005) EMBO J., 24, 2342–2353.
27) Gavard, J. & Gutkind, J.S. (2006) Nat. Cell Biol., 8, 1223–1234.
28) Chen, X.L., Nam, J.O., Jean, C., Lawson, C., Walsh, C.T., Goka, E., Lim, S.T., Tomar, A., Tancioni, I., Uryu, S., Guan, J.L., Acevedo, L.M., Weis, S.M., Cheresh, D.A., & Schlaepfer, D.D. (2012) Dev. Cell, 22, 146–157.
29) Orsenigo, F., Giampietro, C., Ferrari, A., Corada, M., Galaup, A., Sigismund, S., Ristagno, G., Maddaluno, L., Koh, G.Y., Franco, D., Kurtcuoglu, V., Poulikakos, D., Baluk, P., McDonald, D., Grazia, L.M., & Dejana, E. (2012) Nat. Commun., 3, 1208.
30) Wessel, F., Winderlich, M., Holm, M., Frye, M., Rivera-Galdos, R., Vockel, M., Linnepe, R., Ipe, U., Stadtmann, A., Zarbock, A., Nottebaum, A.F., & Vestweber, D. (2014) Nat. Immunol., 15, 223–230.
31) Sidibe, A., Polena, H., Razanajatovo, J., Mannic, T., Chaumontel, N., Bama, S., Marechal, I., Huber, P., Gulino-Debrac, D., Bouillet, L., & Vilgrain, I. (2014) Am. J. Physiol. Heart Circ. Physiol., 307, H448–H454.
32) Baumeister, U., Funke, R., Ebnet, K., Vorschmitt, H., Koch, S., & Vestweber, D. (2005) EMBO J., 24, 1686–1695.
33) Nawroth, R., Poell, G., Ranft, A., Kloep, S., Samulowitz, U., Fachinger, G., Golding, M., Shima, D.T., Deutsch, U., & Vestweber, D. (2002) EMBO J., 21, 4885–4895.
34) Nottebaum, A.F., Cagna, G., Winderlich, M., Gamp, A.C., Linnepe, R., Polaschegg, C., Filippova, K., Lyck, R., Engelhardt, B., Kamenyeva, O., Bixel, M.G., Butz, S., & Vestweber, D. (2008) J. Exp. Med., 205, 2929–2945.
35) Grazia, L.M., Zanetti, A., Corada, M., Takahashi, T., Balconi, G., Breviario, F., Orsenigo, F., Cattelino, A., Kemler, R., Daniel, T.O., & Dejana, E. (2003) J. Cell Biol., 161, 793–804.
36) Sui, X.F., Kiser, T.D., Hyun, S.W., Angelini, D.J., Del Vecchio, R.L., Young, B.A., Hasday, J.D., Romer, L.H., Passaniti, A., Tonks, N.K., & Goldblum, S.E. (2005) Am. J. Pathol., 166, 1247–1258.
37) Ukropec, J.A., Hollinger, M.K., Salva, S.M., & Woolkalis, M.J. (2000) J. Biol. Chem., 275, 5983–5986.
38) Broermann, A., Winderlich, M., Block, H., Frye, M., Rossaint, J., Zarbock, A., Cagna, G., Linnepe, R., Schulte, D., Nottebaum, A.F., & Vestweber, D. (2011) J. Exp. Med., 208, 2393–2401.
39) Millan, J., Cain, R.J., Reglero-Real, N., Bigarella, C., Marcos-Ramiro, B., Fernandez-Martin, L., Correas, I., & Ridley, A.J. (2010) BMC Biol., 8, 11.
40) Oldenburg, J. & de Rooij, J. (2014) Cell Tissue Res., 355, 545–555.
41) Huveneers, S., Oldenburg, J., Spanjaard, E., van der Krogt, G., Grigoriev, I., Akhmanova, A., Rehmann, H., & de Rooij, J. (2012) J. Cell Biol., 196, 641–652.
42) Dorland, Y.L. & Huveneers, S. (2017) Cell. Mol. Life Sci., 74, 279–292.
43) Garcia-Ponce, A., Citalan-Madrid, A.F., Velazquez-Avila, M., Vargas-Robles, H., & Schnoor, M. (2015) Thromb. Haemost., 113, 20–36.
44) Duluc, L. & Wojciak-Stothard, B. (2014) Cell Tissue Res., 355, 675–685.
45) Fukuhra, S., Sakurai, A., Yamagishi, A., Sako, K., & Mochizuki, N. (2006) J. Biochem. Mol. Biol., 39, 132–139.
46) Fukuhara, S., Sakurai, A., Sano, H., Yamagishi, A., Somekawa, S., Takakura, N., Saito, Y., Kangawa, K., & Mochizuki, N. (2005) Mol. Cell. Biol., 25, 136–146.
47) Cullere, X., Shaw, S.K., Andersson, L., Hirahashi, J., Luscinskas, F.W., & Mayadas, T.N. (2005) Blood, 105, 1950–1955.
48) Bos, J.L., de Rooij, J., & Reedquist, K.A. (2001) Nat. Rev. Mol. Cell Biol., 2, 369–377.
49) Bos, J.L. (2005) Curr. Opin. Cell Biol., 17, 123–128.
50) Sakurai, A., Fukuhara, S., Yamagishi, A., Sako, K., Kamioka, Y., Masuda, M., Nakaoka, Y., & Mochizuki, N. (2006) Mol. Biol. Cell, 17, 966–976.
51) Pannekoek, W.J., van Dijk, J.J.G., Chan, O.Y., Huveneers, S., Linnemann, J.R., Spanjaard, E., Brouwer, P.M., van der Meer, A.J., Zwartkruis, F.J.T., Rehmann, H., de Rooij, J., & Bos, J.L. (2011) Cell. Signal., 23, 2056–2064.
52) Chrzanowska-Wodnicka, M., White, G.C. 2nd, Quilliam, L.A., & Whitehead, K.J. (2015) PLoS One, 10, e0145689.
53) Price, L.S., Hajdo-Milasinovic, A., Zhao, J., Zwartkruis, F.J., Collard, J.G., & Bos, J.L. (2004) J. Biol. Chem., 279, 35127–35132.
54) Knox, A.L. & Brown, N.H. (2002) Science, 295, 1285–1288.
55) Hogan, C., Serpente, N., Cogram, P., Hosking, C.R., Bialucha, C.U., Feller, S.M., Braga, V.M., Birchmeier, W., & Fujita, Y. (2004) Mol. Cell. Biol., 24, 6690–6700.
56) Stockton, R.A., Shenkar, R., Awad, I.A., & Ginsberg, M.H. (2010) J. Exp. Med., 207, 881–896.
57) Glading, A., Han, J., Stockton, R.A., & Ginsberg, M.H. (2007) J. Cell Biol., 179, 247–254.
58) Wilson, C.W., Parker, L.H., Hall, C.J., Smyczek, T., Mak, J., Crow, A., Posthuma, G., De Maziere, A., Sagolla, M., Chalouni, C., Vitorino, P., Roose-Girma, M., Warming, S., Klumperman, J., Crosier, P.S., & Ye, W. (2013) Blood, 122, 3678–3690.
59) Post, A., Pannekoek, W.J., Ponsioen, B., Vliem, M.J., & Bos, J.L. (2015) Mol. Cell. Biol., 35, 2495–2502.
60) Ando, K., Fukuhara, S., Moriya, T., Obara, Y., Nakahata, N., & Mochizuki, N. (2013) J. Cell Biol., 202, 901–916.
61) Wakayama, Y., Fukuhara, S., Ando, K., Matsuda, M., & Mochizuki, N. (2015) Dev. Cell, 32, 109–122.
62) Rao, M.V. & Zaidel-Bar, R. (2016) Mol. Biol. Cell, 27, 2844–2856.
63) Phng, L.K., Gebala, V., Bentley, K., Philippides, A., Wacker, A., Mathivet, T., Sauteur, L., Stanchi, F., Belting, H.G., Affolter, M., & Gerhardt, H. (2015) Dev. Cell, 32, 123–132.
64) Barry, D.M., Xu, K., Meadows, S.M., Zheng, Y., Norden, P.R., Davis, G.E., & Cleaver, O. (2015) Development, 142, 3058–3070.
65) Tian, X., Tian, Y., Gawlak, G., Meng, F., Kawasaki, Y., Akiyama, T., & Birukova, A.A. (2015) Mol. Biol. Cell, 26, 636–650.
66) Birukova, A.A., Zagranichnaya, T., Fu, P., Alekseeva, E., Chen, W., Jacobson, J.R., & Birukov, K.G. (2007) Exp. Cell Res., 313, 2504–2520.
67) Arthur, W.T., Quilliam, L.A., & Cooper, J.A. (2004) J. Cell Biol., 167, 111–122.
68) Stockton, R.A., Schaefer, E., & Schwartz, M.A. (2004) J. Biol. Chem., 279, 46621–46630.
69) Boettner, B., Govek, E.E., Cross, J., & Van, A.L. (2000) Proc. Natl. Acad. Sci. USA, 97, 9064–9069.
70) Lafuente, E.M., van Puijenbroek, A.A., Krause, M., Carman, C.V., Freeman, G.J., Berezovskaya, A., Constantine, E., Springer, T.A., Gertler, F.B., & Boussiotis, V.A. (2004) Dev. Cell, 7, 585–595.
71) Adamson, R.H., Ly, J.C., Sarai, R.K., Lenz, J.F., Altangerel, A., Drenckhahn, D., & Curry, F.E. (2008) Am. J. Physiol. Heart Circ. Physiol., 294, H1188–H1196.
72) Birukova, A.A., Meng, F., Tian, Y., Meliton, A., Sarich, N., Quilliam, L.A., & Birukov, K.G. (2015) Biochim. Biophys. Acta, 1852, 778–791.
73) Birukova, A.A., Fu, P., Xing, J., & Birukov, K.G. (2009) J. Appl. Physiol., 107, 1900–1910.
74) McVerry, B.J., Peng, X., Hassoun, P.M., Sammani, S., Simon, B.A., & Garcia, J.G. (2004) Am. J. Respir. Crit. Care Med., 170, 987–993.
75) Abbasi, T. & Garcia, J.G. (2013) Handb. Exp. Pharmacol., 216, 201–226.
76) Mikelis, C.M., Simaan, M., Ando, K., Fukuhara, S., Sakurai, A., Amornphimoltham, P., Masedunskas, A., Weigert, R., Chavakis, T., Adams, R.H., Offermanns, S., Mochizuki, N., Zheng, Y., & Gutkind, J.S. (2015) Nat. Commun., 6, 6725.
77) Gorovoy, M., Neamu, R., Niu, J., Vogel, S., Predescu, D., Miyoshi, J., Takai, Y., Kini, V., Mehta, D., Malik, A.B., & Voyno-Yasenetskaya, T. (2007) Circ. Res., 101, 50–58.
78) Cinel, I., Ark, M., Dellinger, P., Karabacak, T., Tamer, L., Cinel, L., Michael, P., Hussein, S., Parrillo, J.E., Kumar, A., & Kumar, A. (2012) J. Thorac. Dis., 4, 30–39.
79) Fukuhara, S., Sako, K., Noda, K., Zhang, J., Minami, M., & Mochizuki, N. (2010) Histol. Histopathol., 25, 387–396.
80) Thurston, G., Suri, C., Smith, K., McClain, J., Sato, T.N., Yancopoulos, G.D., & McDonald, D.M. (1999) Science, 286, 2511–2514.
81) Gavard, J., Patel, V., & Gutkind, J.S. (2008) Dev. Cell, 14, 25–36.
82) Mammoto, T., Parikh, S.M., Mammoto, A., Gallagher, D., Chan, B., Mostoslavsky, G., Ingber, D.E., & Sukhatme, V.P. (2007) J. Biol. Chem., 282, 23910–23918.
83) Kidoya, H., Ueno, M., Yamada, Y., Mochizuki, N., Nakata, M., Yano, T., Fujii, R., & Takakura, N. (2008) EMBO J., 27, 522–534.
84) Fukuhara, S., Sako, K., Minami, T., Noda, K., Kim, H.Z., Kodama, T., Shibuya, M., Takakura, N., Koh, G.Y., & Mochizuki, N. (2008) Nat. Cell Biol., 10, 513–526.
85) Saharinen, P., Eklund, L., Miettinen, J., Wirkkala, R., Anisimov, A., Winderlich, M., Nottebaum, A., Vestweber, D., Deutsch, U., Koh, G.Y., Olsen, B.R., & Alitalo, K. (2008) Nat. Cell Biol., 10, 527–537.
86) Kim, D.H., Jung, Y.J., Lee, A.S., Lee, S., Kang, K.P., Lee, T.H., Lee, S.Y., Jang, K.Y., Moon, W.S., Choi, K.S., Yoon, K.H., Sung, M.J., Park, S.K., & Kim, W. (2009) Kidney Int., 76, 1180–1191.
87) Joussen, A.M., Poulaki, V., Tsujikawa, A., Qin, W., Qaum, T., Xu, Q., Moromizato, Y., Bursell, S.E., Wiegand, S.J., Rudge, J., Ioffe, E., Yancopoulos, G.D., & Adamis, A.P. (2002) Am. J. Pathol., 160, 1683–1693.
88) Lee, J., Park, D.Y., Park, D.Y., Park, I., Chang, W., Nakaoka, Y., Komuro, I., Yoo, O.J., & Koh, G.Y. (2014) Invest. Ophthalmol. Vis. Sci., 55, 2191–2199.
89) Han, S., Lee, S.J., Kim, K.E., Lee, H.S., Oh, N., Park, I., Ko, E., Oh, S.J., Lee, Y.S., Kim, D., Lee, S., Lee, D.H., Lee, K.H., Chae, S.Y., Lee, J.H., Kim, S.J., Kim, H.C., Kim, S., Kim, S.H., Kim, C., Nakaoka, Y., He, Y., Augustin, H.G., Hu, J., Song, P.H., Kim, Y.I., Kim, P., Kim, I., & Koh, G.Y. (2016) Sci. Transl. Med., 8, 335ra55.
著者紹介Author Profile
 福原 茂朋(ふくはら しげとも)
福原 茂朋(ふくはら しげとも)日本医科大学先端医学研究所病態解析学部門教授.博士(学術).
略歴1996年日本学術振興会・特別研究員(DC2),97年筑波大学大学院博士課程修了,98年米国NIH・ポスドク,2000年熊本大学発生研助手,03年国立循環器病研究センター研究所室員(05年より室長),16年より現職.
研究テーマと抱負体の中の細胞を解析する“in vivo細胞生物学研究”を実践することで,血管形成をはじめとした様々な生命現象を明らかにする.生きた個体の中の細胞の振る舞いを直接目で見て観察していると,思いもよらない現象に出会うことがあり,とてもエキサイティングです.
ウェブサイトhttp://www.nmsbyoutai.com/
趣味スポーツ観戦,ビール.