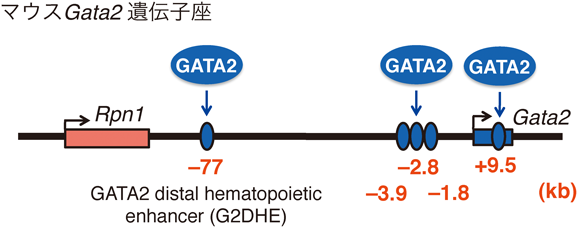
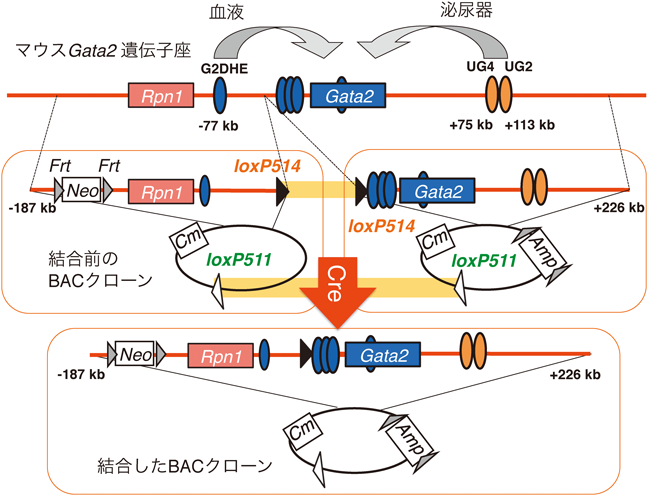
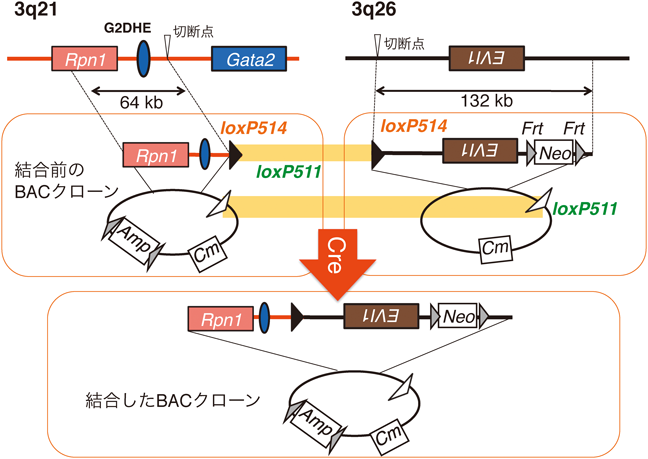
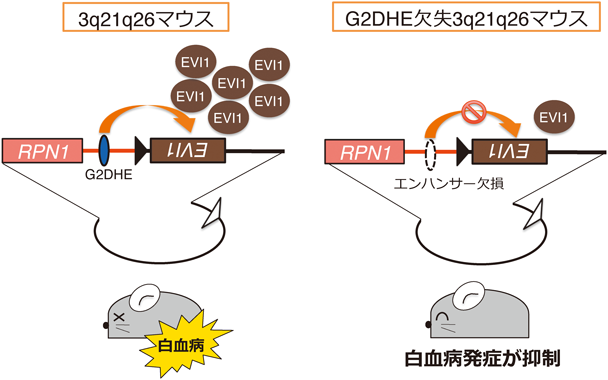
1) Matsuda, M., Sakamoto, N., & Fukumaki, Y. (1992) Blood, 80, 1347–1351.
5) Mansour, M.R., Abraham, B.J., Anders, L., Berezovskaya, A., Gutierrez, A., Durbin, A.D., Etchin, J., Lawton, L., Sallan, S.E., Silverman, L.B., Loh, M.L., Hunger, S.P., Sanda, T., Young, R.A., & Look, A.T. (2014) Science, 346, 1373–1377.
6) De Gobbi, M., Viprakasit, V., Hughes, J.R., Fisher, C., Buckle, V.J., Ayyub, H., Gibbons, R.J., Vernimmen, D., Yoshinaga, Y., de Jong, P., Cheng, J.F., Rubin, E.M., Wood, W.G., Bowden, D., & Higgs, D.R. (2006) Science, 312, 1215–1217.
8) Gröschel, S., Sanders, M.A., Hoogenboezem, R., de Wit, E., Bouwman, B.A., Erpelinck, C., van der Velden, V.H., Havermans, M., Avellino, R., van Lom, K., Rombouts, E.J., van Duin, M., Döhner, K., Beverloo, H.B., Bradner, J.E., Döhner, H., Löwenberg, B., Valk, P.J., Bindels, E.M., de Laat, W., & Delwel, R. (2014) Cell, 157, 369–381.
9) Northcott, P.A., Lee, C., Zichner, T., Stütz, A.M., Erkek, S., Kawauchi, D., Shih, D.J., Hovestadt, V., Zapatka, M., Sturm, D., Jones, D.T., Kool, M., Remke, M., Cavalli, F.M., Zuyderduyn, S., Bader, G.D., VandenBerg, S., Esparza, L.A., Ryzhova, M., Wang, W., Wittmann, A., Stark, S., Sieber, L., Seker-Cin, H., Linke, L., Kratochwil, F., Jäger, N., Buchhalter, I., Imbusch, C.D., Zipprich, G., Raeder, B., Schmidt, S., Diessl, N., Wolf, S., Wiemann, S., Brors, B., Lawerenz, C., Eils, J., Warnatz, H.J., Risch, T., Yaspo, M.L., Weber, U.D., Bartholomae, C.C., von Kalle, C., Turányi, E., Hauser, P., Sanden, E., Darabi, A., Siesjö, P., Sterba, J., Zitterbart, K., Sumerauer, D., van Sluis, P., Versteeg, R., Volckmann, R., Koster, J., Schuhmann, M.U., Ebinger, M., Grimes, H.L., Robinson, G.W., Gajjar, A., Mynarek, M., von Hoff, K., Rutkowski, S., Pietsch, T., Scheurlen, W., Felsberg, J., Reifenberger, G., Kulozik, A.E., von Deimling, A., Witt, O., Eils, R., Gilbertson, R.J., Korshunov, A., Taylor, M.D., Lichter, P., Korbel, J.O., Wechsler-Reya, R.J., & Pfister, S.M. (2014) Nature, 511, 428–434.
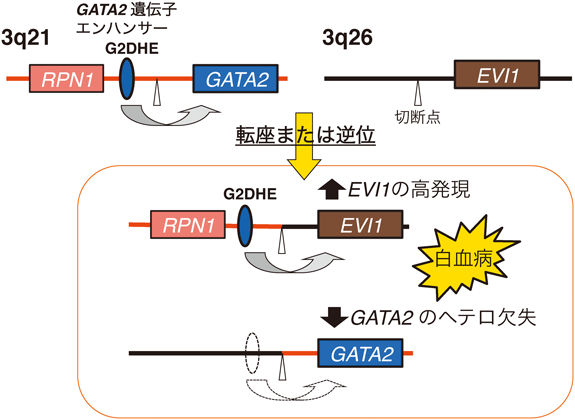
14) Suzuki, M., Kobayashi-Osaki, M., Tsutsumi, S., Pan, X., Ohmori, S., Takai, J., Moriguchi, T., Ohneda, O., Ohneda, K., Shimizu, R., Kanki, Y., Kodama, T., Aburatani, H., & Yamamoto, M. (2013) Genes Cells, 18, 921–933.
29) Dickinson, R.E., Griffin, H., Bigley, V., Reynard, L.N., Hussain, R., Haniffa, M., Lakey, J.H., Rahman, T., Wang, X.N., McGovern, N., Pagan, S., Cookson, S., McDonald, D., Chua, I., Wallis, J., Cant, A., Wright, M., Keavney, B., Chinnery, P.F., Loughlin, J., Hambleton, S., Santibanez-Koref, M., & Collin, M. (2011) Blood, 118, 2656–2658.
30) Hsu, A.P., Sampaio, E.P., Khan, J., Calvo, K.R., Lemieux, J.E., Patel, S.Y., Frucht, D.M., Vinh, D.C., Auth, R.D., Freeman, A.F., Olivier, K.N., Uzel, G., Zerbe, C.S., Spalding, C., Pittaluga, S., Raffeld, M., Kuhns, D.B., Ding, L., Paulson, M.L., Marciano, B.E., Gea-Banacloche, J.C., Orange, J.S., Cuellar-Rodriguez, J., Hickstein, D.D., & Holland, S.M. (2011) Blood, 118, 2653–2655.
31) Ostergaard, P., Simpson, M.A., Connell, F.C., Steward, C.G., Brice, G., Woollard, W.J., Dafou, D., Kilo, T., Smithson, S., Lunt, P., Murday, V.A., Hodgson, S., Keenan, R., Pilz, D.T., Martinez-Corral, I., Makinen, T., Mortimer, P.S., Jeffery, S., Trembath, R.C., & Mansour, S. (2011) Nat. Genet., 43, 929–931.
32) Hahn, C.N., Chong, C.E., Carmichael, C.L., Wilkins, E.J., Brautigan, P.J., Li, X.C., Babic, M., Lin, M., Carmagnac, A., Lee, Y.K., Kok, C.H., Gagliardi, L., Friend, K.L., Ekert, P.G., Butcher, C.M., Brown, A.L., Lewis, I.D., To, L.B., Timms, A.E., Storek, J., Moore, S., Altree, M., Escher, R., Bardy, P.G., Suthers, G.K., D’Andrea, R.J., Horwitz, M.S., & Scott, H.S. (2011) Nat. Genet., 43, 1012–1017.
55) Lugthart, S., Gröschel, S., Beverloo, H.B., Kayser, S., Valk, P.J., van Zelderen-Bhola, S.L., Jan Ossenkoppele, G., Vellenga, E., van den Berg-de Ruiter, E., Schanz, U., Verhoef, G., Vandenberghe, P., Ferrant, A., Köhne, C.H., Pfreundschuh, M., Horst, H.A., Koller, E., von Lilienfeld-Toal, M., Bentz, M., Ganser, A., Schlegelberger, B., Jotterand, M., Krauter, J., Pabst, T., Theobald, M., Schlenk, R.F., Delwel, R., Döhner, K., Löwenberg, B., & Döhner, H. (2010) J. Clin. Oncol., 28, 3890–3898.
60) Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W.B., Fedorov, O., Morse, E.M., Keates, T., Hickman, T.T., Felletar, I., Philpott, M., Munro, S., McKeown, M.R., Wang, Y., Christie, A.L., West, N., Cameron, M.J., Schwartz, B., Heightman, T.D., La Thangue, N., French, C.A., Wiest, O., Kung, A.L., Knapp, S., & Bradner, J.E. (2010) Nature, 468, 1067–1073.
62) Delmore, J.E., Issa, G.C., Lemieux, M.E., Rahl, P.B., Shi, J., Jacobs, H.M., Kastritis, E., Gilpatrick, T., Paranal, R.M., Qi, J., Chesi, M., Schinzel, A.C., McKeown, M.R., Heffernan, T.P., Vakoc, C.R., Bergsagel, P.L., Ghobrial, I.M., Richardson, P.G., Young, R.A., Hahn, W.C., Anderson, K.C., Kung, A.L., Bradner, J.E., & Mitsiades, C.S. (2011) Cell, 146, 904–917.