それは渡米後間もない1992年9月8日火曜日のことだった.「BMPの受容体と思われる遺伝子を単離したのだが,共同研究でノックアウトマウスを作ってもらえないか.」この日本からの国際電話が,その後20年を超える筆者のBMP機能研究のきっかけとなったのであった.当時はBMPのリガンドが次々とクローニングされ,アクティビンなどとともにTGFβスーパーファミリーを形成すること,リガンドの結合活性を利用して細胞表面には分子量の異なるいくつかの結合タンパク質が存在すること(ここではあえて受容体とはいわない)などがわかり始めたころで,どのような受容体が存在し,そこからどのようなシグナル伝達が惹起されるのかについてはまったく見当がつかないという状況であった.その後,遺伝子ターゲティング法の発展やゲノム配列の解析などと相まって,BMPシグナルの多様性と発生生物学における機能的重要性への理解は大いに深まった.本稿では筆者の在米研究生活とオーバーラップするこの歴史的発展を振り返ってみたいと思う.
1)リガンドと受容体
BMPはもともと筋肉内に骨を誘導する生理活性物質として同定,命名された1).精製タンパク標品のペプチド配列をもとに遺伝子がクローニングされ,TGFβスーパーファミリーのメンバーであることがわかった2).歴史的な理由もあって複数の名前がついている遺伝子もあるが,現在BMPという名前で呼ばれるリガンドは14個である(ただし,すべてのBMPと名前のつくリガンドに骨の誘導活性が確認されているわけではない).なお,BMP1はタンパク分解質酵素であるペプチダーゼM12Aの仲間であり,BMPリガンドといった場合にはそこには含めない3).受容体としては1型,2型が同定されており,2型の方が分子量が大きい.1型が2分子,2型が2分子の四量体受容体を作り,リガンドと結合することがシグナル伝達に必要とされている4).BMPリガンドと結合し情報伝達に関わる1型受容体としてBMPR1A, BMPR1B, ACVR1が,2型受容体としてBMPR2, ActR2A, ActR2Bが同定されている.後述するようにBMPR1AのリガンドとしてBMP2, BMP4が,ACVR1のリガンドとしてBMP6, BMP7が結合し,シグナル伝達を行う5, 6).
2)シグナル伝達因子とシグナル抑制因子
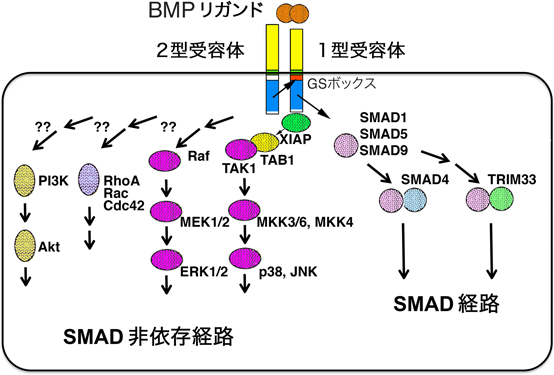
1型,2型受容体ともに細胞膜貫通部位を一つ持ち,また,細胞内部位にはセリントレオニンキナーゼドメインを持つ.リガンドが受容体に結合すると,2型受容体のキナーゼ活性により,1型受容体の膜貫通部位の直下にあるGSボックスと呼ばれる部分がリン酸化されて1型受容体のキナーゼが活性化される.これにより,SMAD1, SMAD5, SMAD9がリン酸化され,次いでSMAD4と複合体を作ることによって核へ移行し,転写活性を制御する.いわゆるBMP-SMADシグナル伝達経路と呼ばれるものである(図1)3).また,SMADを介さず,MAPキナーゼ経路やPI3キナーゼ経路を介するシグナル伝達経路も見つかっており,これらは合わせてSMAD非依存経路と呼ばれている(図1).同じTGFβスーパーファミリーの中でもTGFβやアクティビンの場合にはBMPとは異なる1型,2型受容体の組合わせを必要とし,活性化された1型受容体はSMAD2, SMAD3をリン酸化する.TGFβの場合にはリン酸化されたSMADがTRIM33と結合してSMAD4と結合した場合とは異なる遺伝子発現調節をすることが知られている.BMP-SMADシグナルに対してTRIM33がどのような働きをするのかはまだよくわかっていない.TGFβスーパーファミリーのうち,ミュラー管阻害因子(MIS,抗ミュラー管ホルモン,AMHとも呼ばれる)は特異的に2型受容体AMHR2に結合するが,1型受容体としてはBMPR1AあるいはACVR1を要求し,結果としてSMAD1, SMAD5, SMAD9がリン酸化される7, 8).
細胞外にはBMPリガンドと受容体との結合を阻害する一連のタンパク質群があり,また,細胞内にはリン酸化によって活性化されたSMADの核移行を阻害する阻害的SMAD(SMAD6, SMAD7)があり,これらの発現パターンなどによって発生や細胞機能が詳細に制御されている3, 9, 10).
両生類の胚を用いた実験からBMPや他のTGFβスーパーファミリーの中胚葉誘導への関与が示唆されていた11, 12).両生類ではBMPを過剰発現させることによって腹側中胚葉のマーカー遺伝子が誘導されることなどから,BMPは誘導された中胚葉を腹側へ向けて運命づけると考えられていた13).また,短縮型BMP1A型受容体遺伝子を両生類の胚へ導入して過剰発現させることによってBMPシグナルを遮断させると,背側の中胚葉が増加し,頭部構造の二重化が観察された14).
1)初期発生の仕組み
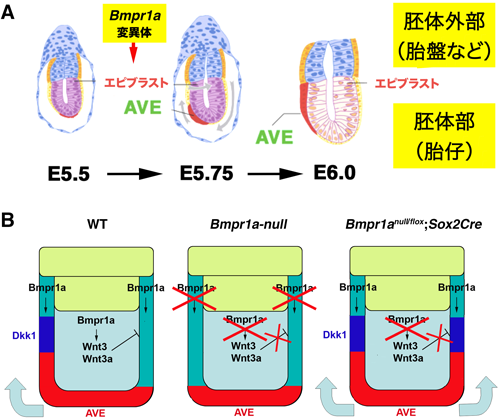
マウス胚では着床の直前に内部細胞塊の位置により一方向の軸(近位遠位)が決まり,近位側は将来背側になる.着床後,遠位側の臓側内胚葉に前方臓側内胚葉(anterior visceral endoderm:AVE)と呼ばれる少数の細胞群が形成され,これら細胞が中胚葉の誘導直前に一方向へ動き出すことが知られている(図2)15–17).AVEが移動した方向と反対側に原条が形成されて胚外胚葉が中胚葉へと誘導されていく.すなわち,AVEが移動した方向が頭側,その反対側が尾側となる.原条が遠位側まで伸びるとそこにノード(結節)が形成され,左右の非対称性が生まれる18, 19).これら初期胚における形態形成,パターニングにBMPシグナルは他の因子と共同して重要な役割を担っている15).
2)BMPリガンドのノックアウトから得られた初期発生への知見
BMPリガンドの遺伝学的解析のうち,初期発生に関連するBMP2, 4, 7について簡単に解説する.ノックアウトによって発生上顕著な異常を示さなかったBMPリガンドについても,その後の解析でさまざまな臓器の恒常性の維持に関与することがわかってきている.
Bmp2のノックアウトではマウス胚は中胚葉誘導の直後に致死となる20).ホモマウスのごく一部において,心臓が腹部側ではなく,背部側に形成される.これは中胚葉誘導前にやはり一部の胚で観察される胚体外の膜組織の融合不全に由来すると思われるが,因果関係は不明である.心臓の異所的な形成が死因のすべてであるような表記をしてある総説が多いが,心臓の拍動機能が必須になるかなり前に致死になること,さらには心臓の異所的形成は129系の純系マウスの一部の胚でしかみられない表現型なので,その表記自体は誤りである.興味深いことに,Bmp2を胎仔部分のみでノックアウトすると胚は全体でノックアウトした場合と似たような発生段階で致死となったが,心臓の異所的な形成は観察されなかった.一方,胚体外でノックアウトした場合には異所的な形成が観察された21).このことは胚体外で産生されたBMP2が初期胚での胚体外の膜の融合や拡張を介して心臓の発生に関わっていることを示唆している.
Bmp4ノックアウトマウスも中胚葉誘導の直後に致死となるが,マウスの系統によって表現型が多少異なり,中胚葉誘導がほとんどみられないものから,誘導後,数日間生存し,生殖細胞の発生に異常を起こすものまでばらつきがある22, 23).
Bmp7ノックアウトマウスは出産時まで発生が続くが腎臓の形成が起きず,出生後致死となる24, 25).下顎骨の融合不全,歯の形成不全も報告されている26–28).
3)受容体のノックアウトから得られたBMPシグナルの初期発生での機能
1型受容体であるBMP1A受容体(BMPR1A,別名ALK3)のノックアウトでは中胚葉誘導がまったく起こらない29).2型受容体であるBmpr2のノックアウトでも同様の表現型となるため30),この二つの受容体から惹起されるBMPシグナルは中胚葉誘導に必須であると結論できる.Bmp2, Bmp4それぞれのノックアウトでは中胚葉誘導は起こるため,一つのリガンドのノックアウトでは他方による相補により表現型が弱くなったことが推察される.
Cre-lox法を用いてSox2-Creと掛け合わせることによりBmpr1aを胎生5日目の胚体部のみでノックアウトすると,約半分の胚には中胚葉の誘導がみられ,背腹のパターン異常を示したが(次節参照),残りの半分は中胚葉誘導がみられなかった31)(図2B).これらの胚ではAVEは胚全体に広がり,明確な前後軸の形成がみられなかった.グローバルノックアウトではAVEは遠位部にとどまり,AVEの動きに関しては逆の表現型となった.AVEは胚体部と胚体外部のつなぎ目付近の頭部側に局所的に発現するDkk1へ向けて移動することが知られている32).グローバルノックアウトではこの発現部位が観察されず,また胚体部特異的ノックアウトではDkk1の発現部位が頭部側に限局されず,リング状に発現していた.これら発現パターンの異常とAVEの移動パターンとが一致するため,中胚葉誘導時のBMPR1Aを介するBMPシグナルの機能の一つとしてDkk1の発現を制御することによりAVEの移動方向を決めていると推測することができる.
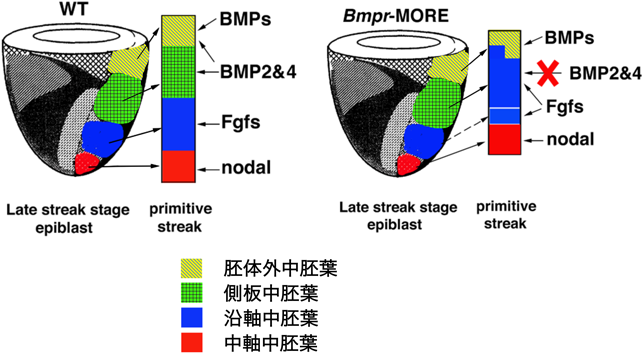
前節で述べたCreよりも1日遅く発現が始まるCre(Meox2-Cre)を用いてBmpr1aを中胚葉誘導の直前に胚体部のみでノックアウトすると中胚葉誘導まで発生が進むが,誘導された中胚葉のパターンに異常が起こる33).前頭部分が肥大し,また通常は左右に一列ずつ形成される体節が複数列形成されるなど,中胚葉が背側化されたと考えらえる形態異常となった.中胚葉誘導時には線維芽細胞増殖因子(fibroblast growth factor:FGF)シグナルが背腹のパターニングに重要であることが知られているが34),Bmpr1a変異胚をFGFシグナルの阻害剤とともに培養するとパターニングの異常が低減したことから,BMPシグナルとFGFシグナルが競合的に働くことで中胚葉誘導時の中胚葉の背腹パターンを制御しているというモデルが提唱できる(図3).
一方,BMPR1Aと構造的に非常によく似ているBMPR1B(別名ALK6)のノックアウトではマウスは致死とならず,四肢の先端部の矮小化,生殖能の消失などの表現型が得られた35–37).これは1A受容体に比べて1B受容体の発現開始時期が遅くまた発現量や発現場所も限局されているためと考えるのが自然であるが,この二つの受容体から惹起されるBMPシグナルのレベルや内容が異なっている可能性も否定できない38).
アクティビン1型受容体(ACVR1,別名ALK2)は当初,TGFβ1型受容体として誤認識,報告され,次いで対合する2型受容体によってアクティビン受容体としてもTGFβ受容体としても働くと誤認識の上塗りをされた不幸な歴史を持つ受容体である(引用文献はわざとつけない).結局これは結合実験でのみ受容体を同定しようとしたところに誤りの原因がある.受容体とは特定のタンパク質に結合し,それによって特異的な情報伝達を行う分子である,という基本概念を忘れたためである.ALK2はTGFβによってもアクティビンによっても特定のシグナルを惹起しない.BMP6やBMP7などのBMPリガンドと結合したときにのみ,BMPR1AやBMPR1Bと同じようなシグナルを惹起する39).したがって,BMP1C型受容体とでも呼ぶべき分子である.しかしながら,前述した不幸な歴史のため,ALK2の正式名称としてアクティビン1型受容体という名前が固定化されてしまい,日々新たな誤解を生み出す結果となっている.本稿ではALK2という呼称を使用する.
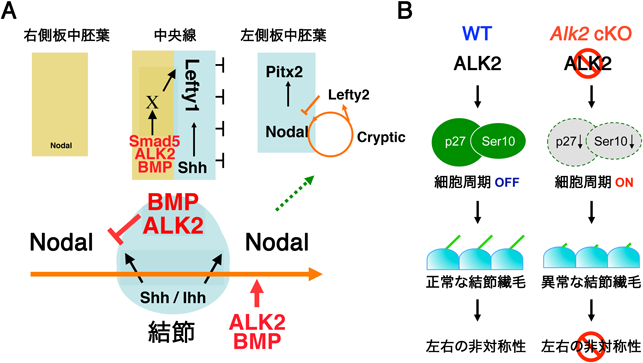
そのAlk2のノックアウトでは中胚葉誘導の途中で発生が止まるという表現型となった40, 41).中胚葉誘導期のマウス胚ではAlk2の発現は胚体外部に限局することから,胚体部でのみAlk2をノックアウトしたところ,予想どおり中胚葉誘導が完了するまで発生は進んだが,左右の非対称性が確立されず,胚の両側が左側として発生した42, 43).マウスなどにおいて左右の非対称性の確立には繊毛の回転によって引き起こされる結節での細胞外液の流れが必要であるとされている19, 44).胚体部特異的Alk2ノックアウトでは繊毛のできが悪く,細胞外液の十分な流れが作り出されない43).繊毛ができるためには細胞の頂端–基底極性ができること,細胞分裂が抑えられて細胞がG0期に入ること,が必要である.マウス胚で結節ができる発生段階ではAlk2は胚体部では結節にのみ発現しており,また結節の細胞のみ分裂が抑えられている42, 45).これはALK2によるBMPシグナルがp27のリン酸化を制御して細胞周期を負に制御するためである43).したがって細胞分裂の盛んな初期胚において,結節でのみALK2が発現し,細胞周期を一時的に止めて繊毛を作ることによって,左右の非対称形成のカスケードが始動するというモデルを考えることができる(図4).
以上述べたように,BMPは初期発生の重要なイベントである三次元的ボディプランの決定にさまざまな観点から関わっていることがわかる.中胚葉誘導前にAVEの移動方向の決定に関わることによって前後軸の決定に,中胚葉誘導時にはFGFシグナルと拮抗することによって背腹のパターニングに,そしてその直後には結節での細胞増殖の制御を介して繊毛運動による左右非対称性の確立にそれぞれ重要であることがわかってきた.ボディプラン確立後の各組織,臓器の発生についてはおびただしい数の論文が発表されている.生殖細胞の分化についてもBMP(特にBMP8A, BMP8B)は必須であるが,受精から着床に至るまでの過程でのBMPの機能についてはまだよくわかっていない.複数のリガンド,受容体からのシグナル,あるいは卵からの持ち込みによってノックアウトの表現型が隠れてしまうことが一因ではないかと思われる46).
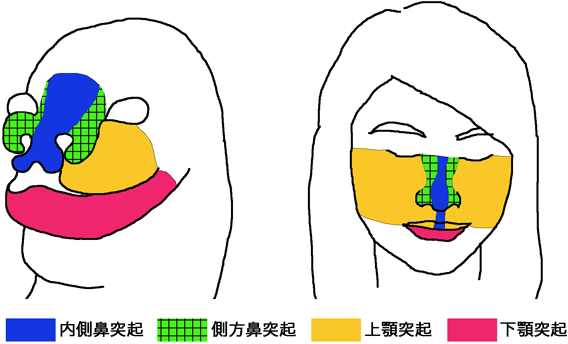
初期胚ではAVEの移動を制御することで前後軸形成への関与が示されているBMPであるが,そのAVEは頭部構造を誘導するオルガナイザーとして重要な働きをしている.頭部はきわめて複雑な発生を行い,したがって細胞分化,組織構築などのプログラムが異常を起こすと先天的異常につながりやすい.実際,口蓋裂など頭部顔面の先天的異常の発症率は非常に高い47).ヒトの場合は胎生5週目ごろ,マウスの場合には10日目ごろに前頭鼻骨隆起が形成される.ここからさらに内側鼻突起,外側鼻突起が一組ずつ形成され,また第一鰓弓から上顎突起,下顎突起が形成される.これらの突起がそれぞれに成長し,特徴的な形をとり,最終的には融合することによって上唇部,鼻腔,上顎,下顎などの構造が作られる(図5).
1)顔面裂と口唇裂
上記に述べた顔面の突起の融合がうまくいかないと顔面裂になる.このうち,内側鼻突起どうし,あるいは内側と外側鼻突起どうしの融合が起こらないと口唇裂になる.次項に述べるように口蓋裂はこれとは異なる仕組みで起こるが,口唇裂と口蓋裂が同時に発症するケースと,口唇裂だけが発症するケースがある.ヒト口唇裂の原因遺伝子としてMSX1, TP63, FGFR1などが同定されている48).Bmpr1aをNestin-Creを用いてノックアウトすると内側鼻突起の細胞死が上昇し両側性完全口唇裂となる49).
また,前頭鼻骨隆起が細胞死の上昇などにより低形成を起こすと,顔の中央に大きな亀裂ができる正中顔面裂になる.前頭鼻骨異形成も前頭鼻骨隆起の低形成が原因と考えられており,ヒトの場合は原因遺伝子としてALX3, ALX4が同定されている.BMPシグナルのアンタゴニストであるNogginとChordinをノックアウトすることにより顔面中央部の組織が低形成になることが報告されており50, 51),BMPシグナル活性と顔面中央部の形成との間に相関があることが示唆される.また,ヘッジホッグシグナルは顔面中央部の発生に重要な働きをしており52),BMPがヘッジホッグシグナルのレベルを制御することで顔面の発生に寄与している可能性も考えられる.
2)口蓋裂
口蓋は,ヒトでは胎生7週目,マウスでは11日目に口蓋棚と呼ばれる一対の組織が舌の左右に形成され,次いで相対的に舌の位置が下がることによって下方を向いていた口蓋棚が互いに向き合うようになり,次いで接触した部分の上皮が細胞死によって消失し,融合が達成される,という過程を経て形成される53).口蓋裂は口腔が大きくならない(したがって舌が移動しない),口蓋棚が低形成で近づかない,口蓋棚が接触するが融合が起きない,などの理由で引き起こされる47).口蓋棚そのものの異常ではなく,舌が移動しないことで二次的に口蓋裂が引き起こされる疾患をピエール・ロバン症候群と呼んでいる.口蓋裂が単独で起きるケースもあるし,前述のように口蓋裂と口唇裂が同時に起きるケースもある.TGFβスーパーファミリーの中ではTgfβ3のノックアウトマウスが口蓋裂を引き起こすことが最初に報告された54).またTGFBの1型受容体であるTgfbr1(別名ALK5)を間葉特異的あるいは上皮特異的にノックアウトした場合にも口蓋裂が起きる55, 56).一方,シグナル因子であるTak1, Smad4, Trim33をそれぞれ単独に上皮特異的にノックアウトした場合には口蓋裂は起きない.またこれらの組合わせで3種類の二重ノックアウトを作製しても口蓋裂は起こらず,Smad4とTrim33の二重ノックアウトをTAK1阻害剤で処理することで初めて口蓋裂が引き起こされた57).これらのことはTGFβがさまざまな下流因子を使って口蓋棚の融合過程を制御していることを示している.
一方,Tak1の神経堤特異的ノックアウトでは口蓋裂が観察された58).11日胚を取り出して下顎と舌を除去して培養すると口蓋棚の融合がみられたため,口蓋裂は下顎の発育不全によって舌が下方に移動しなかったためと推測された58).これはピエール・ロバン症候群の動物モデルとして興味深い.
BMPリガンドの多くやBMPによって発現が制御されるMsx1やMsx2は口蓋棚に多く発現しており,口蓋裂との関連が示唆されていた.実際,Msx1のノックアウトでは口蓋裂が観察される59).Alk2の神経堤特異堤ノックアウトでも口蓋裂が観察されるが,下顎の低形成も同時に起きているので,口蓋棚への直接の影響なのか,ピエール・ロバン症候群様の機構なのかは検討が必要である60).Bmpr1aの神経堤特異的ノックアウトでは心臓の弁機能に異常が出て口蓋形成よりも早い時期に致死となってしまう61, 62).心機能の低下をアドレナリン作動薬で薬理学的に補ってやると出生時まで発生が進み,口蓋裂を含む頭部顔面形成異常を示した63, 64).前述のNestin-Creを用いたBmpr1aのノックアウトでも口蓋裂が観察される49).これらの結果はBMPシグナルが口蓋棚の成長と融合に重要であることを示している.
3)頭蓋癒合症とは
哺乳類の頭蓋や顔面は20数個からの骨で作られている.頭蓋骨は間葉から直接分化してきた骨芽細胞による膜性骨化,頭蓋基底は間葉からまず軟骨細胞が分化し,ついで軟骨が骨に置き換わる内軟骨性骨化により形成される.頭蓋の前半分とほとんどの顔面の骨は神経堤細胞由来であり,頭蓋の後ろ半分は沿軸中胚葉由来である65–67).神経堤細胞由来の骨は中胚葉由来の骨に比べて増殖能が高く,FGFシグナル活性が高い68).頭蓋や顔面の骨どうしは頭蓋縫合と呼ばれる軟組織でつながっており,頭蓋縫合は頭や顔の形を保つとともに,それぞれの骨の増殖をつかさどる組織として重要な働きを果たしている69, 70).頭蓋縫合には未分化な細胞が存在し,そこから骨芽細胞が常に分化している.いったん骨ができると骨としての成長は非常に遅い.頭蓋縫合から常に新しい骨芽細胞が供給され,これらの細胞が既存の骨の端にさらに新しく骨を継ぎ足すことによって,頭蓋,顔面の骨は脳の成長に見合った速度で大きさを増していく.脳の大きさが十分に大きくなるころには頭蓋縫合はだんだんと消失し,成人ではほとんどみられなくなる.骨どうしが癒合するため,成人の骨の数は乳幼児のそれよりも少なくなるというのは知っていると自慢できるトリビアかもしれない.
その頭蓋縫合が何らかの理由で早い時期に消失してしまうと,頭蓋の成長がきわめて遅くなる.一方,脳は成長を続けようとするから,頭蓋内の圧力が上昇し,これによって頭や顔の形にゆがみがでてくる.これが頭蓋癒合症と呼ばれる疾患で,罹患率は2500人に1人と先天性異常の中では非常に高い部類になる.放置すると慢性的な頭痛,視覚聴覚などへの影響がでてくる.現在有効な治療法は癒合部分を取り除き,二つの骨をある程度の距離をおいて固定するという外科的な方法のみである71, 72).しかしながら,しばらく経つと再び癒合が起き,罹患児は十分脳が発達する年齢になるまで場合によっては数回の外科手術を受ける必要がある.
遺伝学的手法やゲノムワイド解析などにより,これまで100を超える原因遺伝子が同定されているが,それでも全症例の30%程度にしかならない73, 74).原因遺伝子が異なれば発症機構も異なり,したがって有効な治療法も変わってくることが予測される.原因遺伝子のさらなる検索,相応する動物モデルの創出,そして薬剤スクリーニングを効率的に行えるような試験管内システムの構築が急務となる.
4)頭蓋癒合症とBMP
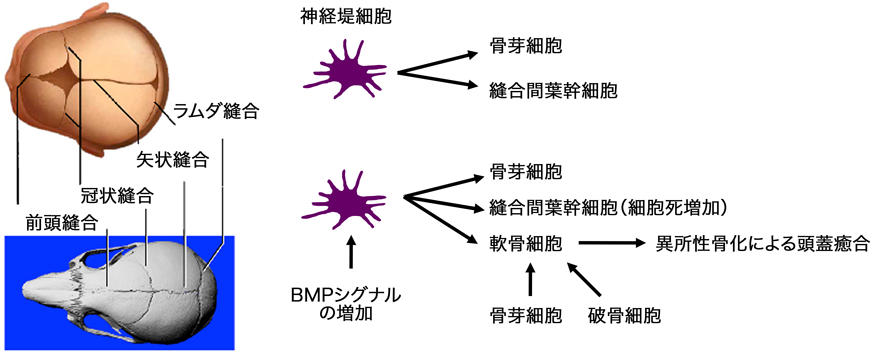
頭蓋発生へのBMPの関与はいくつか知られている.また,ボストン型頭蓋癒合症の原因遺伝子としてBMPシグナルによって発現が正に制御されるMSX2が同定されていること75, 76),BMP抑制因子であるNOGGINが頭蓋縫合に強く発現しており,またNOGGIN強制発現によって頭蓋縫合の癒合が抑えられること77)から,BMPシグナルの頭蓋癒合症への関わりが予測されていた.前述したように1型受容体にはキナーゼ活性を制御するGSボックスと呼ばれるドメインがあり,BMPR1Aの場合には233番目のグルタミンをアスパラギン酸へ変えるとキナーゼ活性が構成的,すなわち,BMPリガンドと結合せずにBMPシグナル活性を惹起することが知られている(BMPR1A-Q233D).そこでBMPR1A-Q233Dを神経堤細胞特異的に発現させると,左右の前頭骨をつなぐ前頭頭蓋縫合線の消失がみられた19).過剰なBMPシグナルによって骨の増殖が盛んになり,軟組織を埋めつくしてしまった,というのが単純な説明である.ところが骨芽細胞特異的にBMPR1A-Q233Dを発現させても何も変化がない.また,1型受容体キナーゼの阻害剤,LDN193189の妊娠マウスへの投与によって頭蓋癒合を抑えることができるが,頭蓋の骨化が起きる胎生16日からの投与では抑制が起きず,さらに早い段階からの投与が必要であった19, 78).これらのことはすでに骨芽細胞への分化にコミットした細胞ではなく,より未分化な神経堤細胞でBMPシグナルが増加することが,発生が進んでからの頭蓋縫合の消失の原因であるということを示唆している(図6).
それではBMPが神経堤細胞へ及ぼす影響とは何なのであろうか.頭蓋癒合症を起こしたマウスの頭蓋が薄いことから細胞死の増加が想定され,実際にTUNEL陽性の細胞数の増加とp53タンパク質の増加が観察された47, 79).p53の核移行を抑えるPifithrinを投与すると表現型の大幅な改善がみられ47, 79),また頭蓋縫合も癒合せずに保たれた(未発表).これらの結果は過剰なBMPシグナルによって頭蓋縫合部にある前駆細胞の細胞死が亢進して枯渇することによって癒合が起こるという機構を示唆している.頭蓋縫合ではGli1の発現が高い80, 81).Gli1を発現している細胞を特異的に除去すると多くの頭蓋縫合が消失する80).また,神経堤でのBMP過剰発現マウスでは影響を受ける頭蓋縫合部分でのみ,Gli1の発現が消失していた(未発表).これらのことから,頭蓋縫合にはGli1でマークされる幹細胞群が存在し,そこから常に骨芽細胞が分化してくることで,頭蓋縫合の維持と頭蓋骨の成長が起きていると考えることができる(図6).
それでは実際にBMPシグナルが増強したことで頭蓋癒合症が起きたケースはあるのだろうか.現在までのところ,BMPR1Aの変異が発症原因であるという報告はない.受容体そのものに変異によるアミノ酸置換が起きて活性が変化するとおそらく胎生致死になると考えられる82).発現制御部位の変異で組織特異的,あるいは時期特異的に発現が変化したような場合にのみ,致死をまぬがれ,なおかつ異常を引き起こすといったケースが可能と思われる.このためにはエクソームだけでなくゲノム全体の解析が必要となるため,まだ発見されずにいると考えたほうがよいと思われる.非症候群性の前頭および矢状縫合早期癒合症のゲノム解析からBMP2遺伝子に近い場所にある一塩基多型(SNP)との相関が見いだされている83).この部分がエンハンサーとして働き,BMP2の発現を頭蓋特異的に上昇させているのではという予測がなされている.また,別のコホートの解析からこのBMP2部位のSNPとともにSMAD6に変異を持つ患者はすべて非症候群性の前頭および矢状縫合早期癒合症を発症していることが報告された84).SMAD6はBMPシグナルを抑制的に制御しているので,SNPによるBMP2の発現上昇と,抑制の消失の両方が同時に起きることでBMPシグナルがさらに活性化されて頭蓋癒合症になるというモデルが考えられる85, 86).
ヒトと異なりマウスの場合,ほとんどの頭蓋縫合は成長しても消失しないが,前頭縫合の後方部は生後40日程度までに消失する.消失の前に縫合内の軟組織の部分に軟骨ができることが知られている68).一方,FGFシグナルの過剰発現で前頭骨と頭頂骨をつなぐ冠状縫合が消失するApert症候群やCrouzon症候群においても,消失の直前に軟骨ができることが知られている87).FGFシグナルとWntシグナルが同時に上昇したマウスでは頭頂骨どうしをつなぐ矢状縫合が消失するが,やはり消失の前に軟骨組織が観察される88).これらの状況証拠から想像力を働かせると,頭蓋の骨は膜性骨化を経て形成されるが縫合が消失するときは内軟骨性骨化が起きていると考えることができる.これまで頭蓋癒合症は骨の疾患,骨のできすぎによる疾患と思われていたが,幹細胞の細胞系譜決定の異常により異所性軟骨の形成で誘起される「あってはならない場所」での内軟骨性骨化で引き起こされる疾患であると捉え直すことができるかもしれない.次節で述べるように,BMPは幹細胞に働きかけて軟骨方向へと分化を誘導する力がある.BMPの過剰発現で頭蓋癒合症が起きる場合にも同様の機構で説明できる可能性がある(図6).
5. 骨の発生,恒常性維持への関与:50年来の夢から醒めるとき
骨を作る因子ということで発見,命名されたBMPである.骨を作るということで,臨床応用が期待され,実際に欧米では整形外科領域での骨折治癒に使われている.しかしながら,我々のBMPと骨についての理解はそれでよいのであろうか.実際に,ヒト血中に観察されるBMPの濃度は300~600 pg/mLであり,細胞培養時にBMPシグナルを惹起させる濃度は100 ng/mL程度である.ところが治療のために骨折部位の骨膜細胞をターゲットに投与されるBMP2やBMP7はミリグラムの単位を必要とする89–91).すなわち,期待される生理活性を導くのに実はとんでもない量が必要とされていることになる.実際,細胞培養系で2000 ng/mLの高濃度でBMP2を添加すると骨膜細胞の細胞増殖が抑制され,骨形成には負の結果を誘導することが推測された92).これは,TGFβが濃度依存的に反対の生理的効果を発揮する現象に類似している93).また,現在の臨床濃度では期待された骨形成効果が得られないという臨床成績も報告されている94, 95).より効果的な臨床応用を目指して,BMPリガンドに変異を導入する,アクティビンとの間でドメインどうしの交換を行うなどの改変を行い,受容体への親和性を上げる,組織中での分解を防ぐ,といった工夫はされているが96, 97),BMPの骨格系への作用は骨芽細胞に作用して造骨活性を上げるというこれまでの単純な図式では説明できない.
1)骨格系特異的ノックアウト
骨格系で発現量の高いBmp2, Bmp4, Bmpr1aのストレートノックアウトはいずれも初期胚で致死となる.Cre-loxを使った組織特異的ノックアウトが開発されると,さっそくこれをとり入れた骨芽細胞,あるいは軟骨細胞に特異的なノックアウトマウスが作製された.BMPシグナルが骨芽細胞の増殖分化に必須であるのなら,骨格系がそもそも発生してこないことも含めた骨格形成異常,骨量の低下などの表現型が期待できる.しかしながら,初期の報告は予測をほどよく裏切るものであった98).
2)リガンドのノックアウト:BMPは骨形成因子ではなく骨折治癒因子なのか
骨芽細胞は未分化な間葉から分化してくるが,Bmp2とBmp4をともにPrx1-Creを用いて間葉特異的にノックアウトすると軟骨形成が悪くなり,したがって手足の骨の形成が非常に抑えられる99).Bmp2, Bmp4, Bmp7それぞれの単独のノックアウトでは大きな変化はみられず,またBmp2とBmp7との二重ノックアウトでも大きな変化はみられなかった99).一方,Bmp2とBmp4の単独ノックアウトで骨折治癒への影響をみると,Bmp4ノックアウトでは正常であったがBmp2ノックアウトでは治癒が起こらず,BMP2は間葉細胞が骨折治癒に向けて分化するために必須であることが示された100).
BMP4を1型コラーゲンプロモーターを用いて骨芽細胞特異的に過剰生産させると,骨量が増すという予測に反して,骨量の大幅な減少が観察された101).破骨細胞の増加がみられたことから,BMPが破骨細胞の分化に関与している可能性が示唆された.ここから紡ぎ出される議論については次項で詳しく述べる.
3)BMP受容体のノックアウト:BMPは骨形成因子ではなく骨調節因子なのか
1型受容体であるBMP1A受容体(Bmpr1a)はいくつかの異なる骨特異的なCreマウスを用いてノックアウトされている.骨芽細胞の後期分化マーカーであるオステオカルシンのプロモーターでCreを発現させると(Og2-Cre),マウスの成長がやや遅くなり,生後1か月では骨量のわずかな減少がみられた.ところがこれらマウスを生後6か月で観察すると変異マウスの骨量が逆に増加することが観察された102).Og2-CreによるSmad4のノックアウトでも週齢に伴う表現型の遷移がみられた103).また,未分化幹細胞に選択的に発現するMx1-Creを用いてBmpr1aをノックアウトすると海綿骨の増加がみられたが,Cre活性を誘導する時期(生後1週間と3週間)によって長管骨内での海綿骨の増加する場所に違いがみられた104).これらのことはIA型受容体を介するBMPシグナルの骨芽細胞での生理的機能がマウスの週齢によって変化することを示唆している.しかしながら,タモキシフェンで活性が誘導される1型コラーゲンプロモーター(Col1-CreERT)による骨芽細胞特異的なCreを使った系では胎発生期,離乳期,成熟後のいずれの時期でも変異マウスの方が骨量が多く,骨密度も高いという結果になった105, 106).1型コラーゲンは骨芽細胞の分化段階としてはオステオカルシンより早い時期に発現が始まるので,BMPの骨芽細胞での機能はマウスの週齢ばかりでなく,骨芽細胞の分化段階によっても異なる可能性がある.
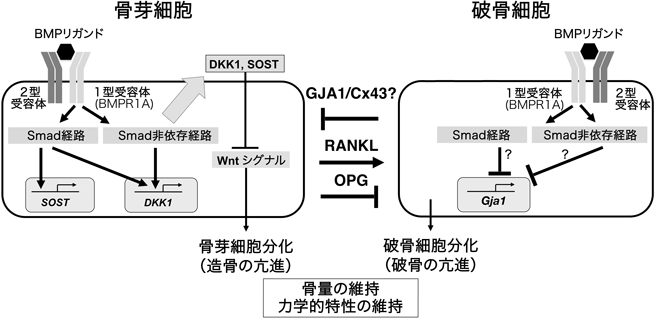
ではなぜ予想に反して骨量が増えたのであろうか.カルセインによる二重ラベル法でミネラルの沈着速度を調べると変異マウスの方で有意な低下がみられた.同時に骨中の破骨細胞の数が非常に低下していることが観察された.これらのことから骨芽細胞におけるBMPシグナルは骨芽細胞による増骨作用と骨芽細胞による破骨細胞の分化増殖との両方に関わっており,骨芽細胞でBMPシグナルを遮断することにより造骨,破骨ともに低下するが,Bmpr1aをCol1-CreERTでノックアウトした場合には破骨の低下の方がより大きく,結果として骨量が増えるというモデルが考えられた106, 107)(図7).
破骨細胞は単球系から分化してくる骨の基質の分解に関わる多核細胞である.その分化には骨芽細胞の存在が必須であることが知られている.Og2-CreによるBmpr1aのノックアウトの系で破骨細胞の活性が下がっていることが観察されたので,BMPの骨芽細胞での役割の一つに骨芽細胞による破骨細胞の分化の亢進があることが予測された.1型コラーゲンCre(Col1-CreERT)によりBMP1A受容体をノックアウトした骨芽細胞には破骨細胞の分化を誘導する力が非常に弱い.骨芽細胞は破骨細胞の分化を促進するRANKL,抑制するOPGを分泌し,破骨細胞の増殖はそのバランスによって制御されている.変異骨芽細胞ではRANKLが低下,OPGが増加し,結果として破骨細胞の分化が低下していた.これはBMPシグナルの下流にDkk1,スクレロスチンといったWntカノニカルシグナルの抑制因子があり,ノックアウトによってこれら抑制因子の発現が低下することによって骨芽細胞でのWntシグナルが上昇するためである107).これらのことから,骨芽細胞でBMPシグナルを遮断することによって骨芽細胞による骨産生,破骨細胞による骨吸収はともに低下するが,骨吸収の低下の方がより大きいために,結果として骨量が増えたと考えることができる(図7).
また,破骨細胞内でのBMPシグナルは破骨細胞の分化増殖に重要な働きをしている.破骨細胞特異的にBmpr1aをノックアウトすると破骨細胞の分化が抑制されて骨の吸収が減り,その結果骨量の増加が観察される108).ところが同時に骨芽細胞,骨細胞によるミネラルの形成速度が非常に高まるという予測されない結果も得られた108).骨芽細胞,骨細胞でノックアウトが起きていないことは確認されたので,これは破骨細胞から骨芽細胞へBMP依存的に骨芽細胞の分化を抑制するような仕組みがあることを示唆している.培養系を用いた解析の結果,コネクシン43の関与が示唆された109)(図7).
Col1-CreERTによるBmpr1aのノックアウトの系では骨芽細胞の増殖そのものにはっきりした差はみられなかったが,骨芽細胞,骨細胞特異的に発現するDmp1-Creによるノックアウトでは変異骨芽細胞の増殖能が上がっていることが確認された110, 111).これらのことから興味深い二つの知見が浮かび上がってくる.いずれの場合にもカルセイン標識で測定する骨形成速度は低下していた.これは骨細胞によるミネラルの沈着速度をみているので,骨芽細胞による細胞外基質の合成量は変異マウスで上昇している可能性がある.すなわち,ノックアウトでの骨量の増加は骨吸収の低下だけでなく,骨基質合成の上昇にもよる可能性がある.もう一つは細胞外基質中に入り込んだ骨細胞の数が非常に多くなっていることである.教科書的には骨芽細胞は細胞周期から抜け出し,基質中に入り込んで骨細胞へと最終分化をしていくことになっている.BMPシグナルが遮断されたことによって,骨細胞へと分化する骨芽細胞の割合が増えたという可能性とともに,変異骨細胞は骨基質中で分裂を続けているという可能性も考えられる.初期胚でACVR1を介するBMPシグナルを遮断したときに結節の細胞分裂が止まらず左右の非対称性に異常がでたこと(図4)を考えると,BMPシグナルの持つ機能の一面として興味深い仮説である.
4)BMPと骨の質,BMP飽食の時代
それでは骨芽細胞でのBMPシグナルの遮断によって増えた骨は「よい骨」なのだろうか.それとも「でき損いの骨」なのだろうか.ここでもまた我々の予測は裏切られることになる.歴史的には骨の量が骨の強さの指標として使われてきており,実際,骨粗鬆症の診断基準として骨量の低下が使われている.ところが骨の量だけでは骨折リスクの予想に不十分であることがわかり,量だけではなく骨の「質」も同時に考慮することが必要であるということになってきている.さて,ここでの質であるが,骨の持つ属性のうち,「量」以外のすべてのもの,という定義になっており,分野や研究者によってその内容が同一でないことがある.ここではコラーゲンの性質とミネラルの性質を骨の質として言及し,骨の力学的特性について議論する.コラーゲンは骨芽細胞で産生されたあと,さまざまな修飾を受ける.分泌されたコラーゲンのペプチド鎖どうしの間で,最初は2本のペプチドをつなぐような形で架橋が形成され(二価),次いでそれが3本のペプチドをつなぐ形に変化する(三価).二価の架橋は未成熟な架橋,三価の架橋は成熟した架橋とも呼ばれており,架橋の程度や割合が骨の柔軟性に影響すると考えられている.ミネラルは細胞外基質の中に結晶として蓄えられていくが,この結晶の大きさがそろっているかどうか(結晶化度,クリスタリニティ)が,骨の質の一つとして評価の対象となる.また,ミネラルとコラーゲンを含むタンパク質との比率やミネラルの量そのものが骨の硬さに影響すると考えられている.
前述したようにBmp2のノックアウトでは骨折の増加が観察されている.骨量の顕著な低下が報告されておりこれが理由であると想像されるが,骨の質の変化については解析がされていない.Bmpr1aの骨芽細胞特異的ノックアウトについては骨全体でのコラーゲンの架橋を調べると成熟型のものが増えていた112).骨形成,骨吸収ともに低下しており,骨の代謝回転速度が低下しているために,コラーゲンの架橋が二価から三価へ進む時間が増えたためと考えられる.ラマン分光法を用い,大腿骨の皮質骨部分と海綿骨部分を別々に測定すると海綿骨ではコラーゲン架橋の成熟度,ミネラル・基質比いずれも高かったが,皮質骨部分ではどちらも対照群と変化がなかった.ナノインデンテンション法で力学的特性を調べると,硬度,伸縮性どちらも海綿骨部分では対照群よりも高い値が得られ,皮質骨部分では対照群と同程度であった.これらのことから,BMP遮断によって引き起こされる骨の質の変化,すなわちコラーゲン架橋の成熟度,ミネラル・基質比の上昇が硬度,伸縮度の上昇に寄与している可能性が示された.これらのことはBMPシグナルを遮断することによって骨量が増え,増えた骨は力学的特性上も有利であることを示している.いずれにしても,BMPがない,あるいは少ないほうが骨量も上がり,力学的特性もよくなるということは,我々の骨格系には十分すぎる量のBMPがあり,場合によってはそれを低下させることが骨格系疾患の治療に有効であるのかもしれない.
6. 異所性骨化とBMPシグナル,アクティビンの逆襲
骨格系に関わる疾患としては,発生時の異常によって形態や機能が異常となるもの,造骨と破骨のバランスがくずれて骨量が異常となるものなどがあげられる.この他に,本来の骨格以外の組織に骨ができる異所性骨化と呼ばれるものがある.たとえば人工股関節全置換術を受けた患者の実に90%が人工関節の周りの軟組織に骨様の構造が形成されることが報告されている113, 114).また,重度の火傷を負った患者の60%に,しばらくして火傷した箇所から離れた関節に骨化が起きる115, 116).また,戦場での負傷によって足の切断を余儀なくされた兵士の60%に切断後2か月以内に異所性の骨化が起きる117).後述するように,臨床的には進行性骨化性線維異形成症という疾患があり,不可逆性の異所性骨化を引き起こす病態である.いったん骨ができてしまうと,これを取り除こうとする外科的手術がさらなる骨化の引き金になることが多い118).したがって,異所性骨化の機構を解明し,治療法を見いだすことは臨床的にも急務であり,また異所性骨化の予防と治療は世界各地に兵士を展開している米国国防省の重要な研究テーマの一つでもある.
1)異所性骨化の動物モデル
異所性骨化を起こす動物モデルはいくつか開発されている.コラーゲンなどのキャリアにBMPをまぶし,皮下,あるいは筋肉中に埋め込むというのは50年まえにMarshall R. Uristが行った方法そのものである.また,マウスの背中を60度に加熱することで火傷を導入し,同時にアキレス腱を切断することにより,アキレス腱の周りに異所性骨化を誘導することができる119, 120).この場合は,火傷によって全身性に炎症が起き,ATPが過剰生産されることが異所性骨化の一因となっており,ATP合成阻害剤の投与によって異所性骨化を抑えることができる119).また,特別にデザインされたチャンバーを使ってラットに爆風を与え大腿骨骨折の後に細菌を感染させるという,戦場を再現したような系で異所性骨化を起こすことも行われている121, 122).神経系との関連も注目されており,神経特異的にBMP4を過剰発現するマウスでは四肢の筋肉中に異所性骨化が起こる123, 124).これはBMPにより神経末端からサブスタンスPが放出されるためと考えられている125).また,脳障害によって神経末端からセロトニンが大量放出されることによって異所性骨化が起こることも報告されている126).
前項でも述べたように1型BMP受容体のキナーゼ活性調節部位に点突然変異を入れるとリガンド非依存的にBMP-SMADシグナルを誘導することができる.ALK2の場合には207番目のグルタミンがアスパラギン酸に変わるとキナーゼ活性が構成的になることが知られている(ALK2-Q207D).そこでALK2-Q207DをCre-loxシステムを使ってCre依存的に発現するマウスを作り82),Creを発現するアデノウイルスを筋肉中に打ち込むとその部位に異所性骨化が起きることが確認された127).骨化はBMP受容体のキナーゼ活性の特異的な阻害剤の投与,抗炎症剤の投与,それぞれによって抑えられた127, 128).これらの結果は異所性骨化にはBMPシグナルの亢進とともに炎症が起きることが必要であることを示している.また,レチノイン酸の作動薬は軟骨形成に阻害的であることが知られていたが,これら作動薬がALK2-Q207Dマウスの異所性骨化を抑えることがわかった129).この発見がきっかけとなって次項に述べる進行性骨化性線維異形成症の治療薬としてレチノイン酸作動薬であるパラパルチンの臨床試験が現在進行中である.また,軟骨形成には低酸素状態が必要であることから,低酸素を制御するHIF1の阻害薬の投与が試みられ,予想どおり異所性骨化を抑えることが確認された120, 130).現在,ALK2-Q207Dマウスは多くの研究室で薬剤スクリーニングに使われており,キナーゼ阻害剤をはじめとする異所性骨化抑制にさらに効果的な薬剤が創出されることが期待される.
2)進行性骨化性線維異形成症とBMPシグナル
進行性骨化性線維異形成症(fibrodysplasia ossificans progressiva:FOP)は外傷などがきっかけになって筋肉や関節に異所性骨化が起きるまれな遺伝病である131, 132).発症頻度は200万人に1人といわれており,日本では2007年に国指定の難病として認定された(指定難病272).骨ができるということからBMPとの関連が推測され,最終的にALK2の点突然変異が原因であることがつきとめられた133).現在までのところ,調べられた患者のすべてにALK2の突然変異が確認されている131, 134).最初に報告されたのは206番目のアミノ酸がアルギニンからヒスチジンに変わるR206H変異であるが(ALK2-R206H),この変異が98%以上を占めている133, 135).前項で述べた異所性骨化を起こすALK2-Q207Dマウスと同じ遺伝子,一つ隣りのアミノ酸の変異である.発症のメカニズムははたして同じなのであろうか.
まず,ALK2ローカスにR206H変異をノックインしたマウスの作製が試みられた136).キメラマウスは外傷刺激により異所性骨化を引き起こし,病態モデルマウスとしての正当性が示された.しかしながらR206H変異を持ったヘテロマウスは不明の理由により致死となった.そこでCre-loxを利用してCreが発現した場合にのみ遺伝子座の一部が逆転してR206H変異を含むエクソンが転写されるというマウスが作り出された137).生後にCre活性を全身で誘導すると,数週間の間に大腿部や脊椎骨の周りに異所性骨化がみられた.予想どおり,変異に伴ってBMP-SMADシグナル活性が上昇していたが,興味深いことにALK2-R206Hはアクティビンが結合したときにもBMP型のSMADをリン酸化することが認められた137, 138).抗アクティビン抗体の投与によりこのマウスモデルでの異所性骨化が抑制されることから,R206H変異によりALK2の受容体としてのリガンドの特異性が広がることが進行性骨化性線維異形成症の病因の本質であると考えられる137, 138).この発見を受けて,現在抗アクティビン抗体の臨床試験が行われている.前項で述べたレチノイン酸作動薬パラパルチンの効果もALK2-R206Hマウスで確認されており139),両者の臨床試験の結果に期待が持たれている140).
最初にも述べたように,ALK2はかつてアクティビン受容体として誤って報告された「不幸な歴史」を持つ.アクティビンは他の1型受容体に結合し,TGFβ型のSMADを活性化する.アクティビンはALK2にも結合するがそれによって下流のシグナル活性は惹起されない.不幸なことにいまでも正式名称はアクティビン受容体である.その受容体がアミノ酸一つ変わるだけでアクティビンに結合し,かつBMP型のSMADを活性化する,というのはシグナル伝達の特異性から興味深い発見であるとともに進行性骨化性線維異形成症治療の決定打にもつながる成果である.その不幸な命名に一矢報いたということになるのであろうか.ALK2がBMP受容体として機能すると同時にアクティビンの結合タンパク質としてアクティビンとアクティビン受容体との結合に競合的に働いていると考えると,Alk2ノックアウトマウスの表現型はBMPシグナルの遮断によるばかりでなく,アクティビンシグナルのオーバーロードによる部分もあるのではないか,と妄想はつきない.
1992年に当時北海道大学におられた上野直人博士から国際電話がかかってきて,はや四半世紀がたとうとしている.これだけ長い間一つの遺伝子について研究を続けることになるとは思わなかったがやればやるほど味のでる,スルメのような遺伝子である.そして,この25年はいい意味で予測を裏切られ続けた25年でもあった.骨芽細胞特異的にBMPを遮断して骨が増えるというのはまったく予測していなかった結果であり,サンプルの順番を間違ったのではないかと何度も確認をした.頭蓋癒合症モデルは実はまったく異なるゴールを設定しての実験だった.ケージの中に鼻の短いキュートなマウスを発見したとき,一体どうしてくれたものだろうかとしばらく立ちつくしたこともある.BMPと血管新生との関連を調べたいとマウスを送ったPaul Yuからしばらして電話がかかってきて,異所性骨化が起きた,どうしよう,と意見交換をしたのも懐かしい思い出である.こういった流れを振り返ってつれづれ思うのは,BMPの正体は当初Marshall R. Uristが考えたような骨を作る因子ではなく,未分化幹細胞に働きかけて分化系譜の決定に関わる因子であるということである.名前をつけかえるのであればボディモルフォジェネティックプロテインであり,ボーンモデュレーティングプロテインである.ただ,そんなことよりACVR1をBMPR1Cと改名させたいというのが筆者の届かぬ願いではある.筆者らが作製したALK2-Q207Dが進行性骨化性線維異形成症の疾患モデルマウスとして多くの研究者や製薬会社に利用していただいていることもあって,ここ数年は国際進行性骨化性線維異形成症協会の主催するミーティングにも呼ばれて患者さんやその御家族と交流することができた.この人たちが以前の生活に戻れるような治療法を開発したいという思いを新たにできる機会であった.Marshall R. Uristの業績をたたえ継承するために始まった国際BMP学会は次回の2018年で12回目となる.どのような新しい知見が学会開催地東京で展開されるのか,今から楽しみである.
謝辞Acknowledgments
BMPの世界に入り込むきっかけとなった国際電話の主,上野直人博士(基礎生物学研究所)には幾千万の言葉をつくしても感謝を伝えきれません.当時上野研究室におられた鈴木厚博士(広島大学),西松伸一郎博士(川崎医科大学)とBMPの機能やマウスの表現型について議論を交わしたのも懐かしい思い出です.1998年に自分の研究室を持ってからはたくさんの方々と一緒に研究をする機会に恵まれました.今回の総説で述べた内容に大きく寄与していただいた方々に感謝します(敬称略).岸上哲士(山梨大学),三浦成人(蘇州森茂診療所),福田智一(岩手大学),神谷宣弘(天理大学),小松義広(テキサス大学),Ajeet Singh(ノースカロライナ大学),Yanshuai Zhang(北京教育機構),井浦文香(神奈川県立がんセンター),Honghao Zhang(ミシガン大学),早野暁(岡山大学),Xia Liu(吉林大学),Ce Shi(吉林大学),戸田雅子(福岡歯科大学).加えて,現在一緒に研究活動を続けている19人のチームメンバーに感謝します.また,世界中の多くの研究者との共同研究に恵まれました.すべてを紹介するわけにはいきませんが,特に以下の研究室とは長年にわたる共同研究で多くの成果を上げることができました.Drs. Jian Feng (Texas A&M), Steve Harris (U Texas San Antonio), John Klingensmith (Duke U), Rob Maxson (U South California), Karen Lyons (UCLA), Paul Yu (Harvard), Mitsuo Yamauchi (U North Carolina), Yoshiyuki Mochida (Boston U), Hank Kronenberg (Harvard), Masahiro Iwamoto (U Maryland), Jun Ninomiya-Tsuji (North Carolina State U), Hongxiang Liu (U Georgia Athens), Hongchen Sun (Jilin U), Charlotte Mistretta (U Michigan), David Kohn (U Michigan), Mike Morris (U Michigan), Vesa Kaartinen (U Michigan), Peter Ma (U Michigan), Benjamin Allen (U Michigan), Paul Krebsbach (U Michigan/UCLA), Benjamin Levi (U Michigan).また,本稿の査読をしていただいた神谷宣弘,小松義広,早野暁,大見真衣子,関P. 玲子,齊藤M. まりの各博士に感謝します.最後になりましたが,渡米後に発生生物学とアメリカでの生存術の奥義を伝授いただいたポスドクメンターであるRichard Behringer博士,サイエンスのなんたるかを基礎から叩き込んでいただいた大学院指導教官の帯刀益夫博士,そして高校時代に分子生物学のもたらす興奮を惜しみなく与えていただいた中山伊佐男先生に深く感謝いたします.
ファンディングソース
米国国立衛生科学研究所,米国国防省,国際進行性骨化性線維異形成症協会,理化学研究所,日本学術振興会,ミシガン大学Mキューブ,デルタデンタル協会.
引用文献References
1) Urist, M.R. (1965) Science, 150, 893–899.
2) Wozney, J.M., Rosen, V., Celeste, A.J., Mitsock, L.M., Whitters, M.J., Kriz, R.W., Hewick, R.M., & Wang, E.A. (1988) Science, 242, 1528–1534.
3) Wu, M., Chen, G., & Li, Y.P. (2016) Bone Res., 4, 16009.
4) Shi, Y. & Massague, J. (2003) Cell, 113, 685–700.
5) Kishigami, S. & Mishina, Y. (2005) Cytokine Growth Factor Rev., 16, 265–278.
6) Itoh, F., Watabe, T., & Miyazono, K. (2014) Semin. Cell Dev. Biol., 32, 98–106.
7) Mishina, Y., Rey, R., Finegold, M.J., Matzuk, M.M., Josso, N., Cate, R.L., & Behringer, R.R. (1996) Genes Dev., 10, 2577–2587.
8) Jamin, S.P., Arango, N.A., Mishina, Y., Hanks, M.C., & Behringer, R.R. (2002) Nat. Genet., 32, 408–410.
9) Yanagita, M., Oka, M., Watabe, T., Iguchi, H., Niida, A., Takahashi, S., Akiyama, T., Miyazono, K., Yanagisawa, M., & Sakurai, T. (2004) Biochem. Biophys. Res. Commun., 316, 490–500.
10) Mochizuki, T., Miyazaki, H., Hara, T., Furuya, T., Imamura, T., Watabe, T., & Miyazono, K. (2004) J. Biol. Chem., 279, 31568–31574.
11) Ueno, N., Nishimatsu, S., & Murakami, K. (1990) Prog. Growth Factor Res., 2, 113–124.
12) Asashima, M., Nakano, H., Uchiyama, H., Sugino, H., Nakamura, T., Eto, Y., Ejima, D., Nishimatsu, S., Ueno, N., & Kinoshita, K. (1991) Proc. Natl. Acad. Sci. USA, 88, 6511–6514.
13) Bier, E. & De Robertis, E.M. (2015) Science, 348, aaa5838.
14) Suzuki, A., Thies, R.S., Yamaji, N., Song, J.J., Wozney, J.M., Murakami, K., & Ueno, N. (1994) Proc. Natl. Acad. Sci. USA, 91, 10255–10259.
15) Takaoka, K., Yamamoto, M., & Hamada, H. (2011) Nat. Cell Biol., 13, 743–752.
16) Yamamoto, M., Saijoh, Y., Perea-Gomez, A., Shawlot, W., Behringer, R.R., Ang, S.L., Hamada, H., & Meno, C. (2004) Nature, 428, 387–392.
17) Yamamoto, M., Beppu, H., Takaoka, K., Meno, C., Li, E., Miyazono, K., & Hamada, H. (2009) J. Cell Biol., 184, 323–334.
18) Nakamura, T., Mine, N., Nakaguchi, E., Mochizuki, A., Yamamoto, M., Yashiro, K., Meno, C., & Hamada, H. (2006) Dev. Cell, 11, 495–504.
19) Komatsu, Y. & Mishina, Y. (2013) Cell. Mol. Life Sci., 70, 4659–4666.
20) Zhang, H. & Bradley, A. (1996) Development, 122, 2977–2986.
21) Madabhushi, M. & Lacy, E. (2011) Dev. Cell, 21, 907–919.
22) Winnier, G., Blessing, M., Labosky, P.A., & Hogan, B.L. (1995) Genes Dev., 9, 2105–2116.
23) Fujiwara, T., Dunn, N.R., & Hogan, B.L. (2001) Proc. Natl. Acad. Sci. USA, 98, 13739–13744.
24) Luo, G., Hofmann, C., Bronckers, A.L., Sohocki, M., Bradley, A., & Karsenty, G. (1995) Genes Dev., 9, 2808–2820.
25) Dudley, A.T., Lyons, K.M., & Robertson, E.J. (1995) Genes Dev., 9, 2795–2807.
26) Kouskoura, T., Kozlova, A., Alexiou, M., Blumer, S., Zouvelou, V., Katsaros, C., Chiquet, M., Mitsiadis, T.A., & Graf, D. (2013) PLoS ONE, 8, e59463.
27) Wyatt, A.W., Osborne, R.J., Stewart, H., & Ragge, N.K. (2010) Hum. Mutat., 31, 781–787.
28) Wang, X.P., Suomalainen, M., Jorgez, C.J., Matzuk, M.M., Werner, S., & Thesleff, I. (2004) Dev. Cell, 7, 719–730.
29) Mishina, Y., Suzuki, A., Ueno, N., & Behringer, R.R. (1995) Genes Dev., 9, 3027–3037.
30) Beppu, H., Kawabata, M., Hamamoto, T., Chytil, A., Minowa, O., Noda, T., & Miyazono, K. (2000) Dev. Biol., 221, 249–258.
31) Miura, S., Singh, A.P., & Mishina, Y. (2010) Dev. Biol., 341, 246–254.
32) Kimura-Yoshida, C., Nakano, H., Okamura, D., Nakao, K., Yonemura, S., Belo, J.A., Aizawa, S., Matsui, Y., & Matsuo, I. (2005) Dev. Cell, 9, 639–650.
33) Miura, S., Davis, S., Klingensmith, J., & Mishina, Y. (2006) Development, 133, 3767–3775.
34) Yamaguchi, T.P., Harpal, K., Henkemeyer, M., & Rossant, J. (1994) Genes Dev., 8, 3032–3044.
35) Yi, S.E., Daluiski, A., Pederson, R., Rosen, V., & Lyons, K.M. (2000) Development, 127, 621–630.
36) Baur, S.T., Mai, J.J., & Dymecki, S.M. (2000) Development, 127, 605–619.
37) Yi, S.E., LaPolt, P.S., Yoon, B.S., Chen, J.Y., Lu, J.K., & Lyons, K.M. (2001) Proc. Natl. Acad. Sci. USA, 98, 7994–7999.
38) Shi, C., Iura, A., Terajima, M., Liu, F., Lyons, K., Pan, H., Zhang, H., Yamauchi, M., Mishina, Y., & Sun, H. (2016) Sci. Rep., 6, 24256.
39) Yamashita, H., ten Dijke, P., Franzen, P., Miyazono, K., & Heldin, C.H. (1994) J. Biol. Chem., 269, 20172–20178.
40) Mishina, Y., Crombie, R., Bradley, A., & Behringer, R.R. (1999) Dev. Biol., 213, 314–326.
41) Gu, Z., Reynolds, E.M., Song, J., Lei, H., Feijen, A., Yu, L., He, W., MacLaughlin, D.T., van den Eijnden-van Raaij, J., Donahoe, P.K., & Li, E. (1999) Development, 126, 2551–2561.
42) Kishigami, S., Yoshikawa, S., Castranio, T., Okazaki, K., Furuta, Y., & Mishina, Y. (2004) Dev. Biol., 276, 185–193.
43) Komatsu, Y., Kaartinen, V., & Mishina, Y. (2011) Development, 138, 3915–3920.
44) Yoshiba, S., Shiratori, H., Kuo, I.Y., Kawasumi, A., Shinohara, K., Nonaka, S., Asai, Y., Sasaki, G., Belo, J.A., Sasaki, H., Nakai, J., Dworniczak, B., Ehrlich, B.E., Pennekamp, P., & Hamada, H. (2012) Science, 338, 226–231.
45) Bellomo, D., Lander, A., Harragan, I., & Brown, N.A. (1996) Dev. Dyn., 205, 471–485.
46) Reyes de Mochel, N.S., Luong, M., Chiang, M., Javier, A.L., Luu, E., Toshihiko, F., MacGregor, G.R., Cinquin, O., & Cho, K.W. (2015) Dev. Biol., 397, 45–55.
47) Graf, D., Malik, Z., Hayano, S., & Mishina, Y. (2016) Cytokine Growth Factor Rev., 27, 129–139.
48) Dixon, M.J., Marazita, M.L., Beaty, T.H., & Murray, J.C. (2011) Nat. Rev. Genet., 12, 167–178.
49) Liu, W., Sun, X., Braut, A., Mishina, Y., Behringer, R.R., Mina, M., & Martin, J.F. (2005) Development, 132, 1453–1461.
50) Klingensmith, J., Matsui, M., Yang, Y.P., & Anderson, R.M. (2010) Am. J. Med. Genet. C. Semin. Med. Genet., 154C, 43–51.
51) Yang, Y.P., Anderson, R.M., & Klingensmith, J. (2010) Hum. Mol. Genet., 19, 3030–3042.
52) Brugmann, S.A., Cordero, D.R., & Helms, J.A. (2010) Am. J. Med. Genet. A., 152A, 2995–3006.
53) Lane, J. & Kaartinen, V. (2014) Wiley Interdiscip. Rev. Syst. Biol. Med., 6, 271–278.
54) Kaartinen, V., Voncken, J.W., Shuler, C., Warburton, D., Bu, D., Heisterkamp, N., & Groffen, J. (1995) Nat. Genet., 11, 415–421.
55) Dudas, M., Kim, J., Li, W.Y., Nagy, A., Larsson, J., Karlsson, S., Chai, Y., & Kaartinen, V. (2006) Dev. Biol., 296, 298–314.
56) Dudas, M., Li, W.Y., Kim, J., Yang, A., & Kaartinen, V. (2007) Acta Histochem., 109, 1–14.
57) Lane, J., Yumoto, K., Azhar, M., Ninomiya-Tsuji, J., Inagaki, M., Hu, Y., Deng, C.X., Kim, J., Mishina, Y., & Kaartinen, V. (2015) Dev. Biol., 398, 231–241.
58) Yumoto, K., Thomas, P.S., Lane, J., Matsuzaki, K., Inagaki, M., Ninomiya-Tsuji, J., Scott, G.J., Ray, M.K., Ishii, M., Maxson, R., Mishina, Y., & Kaartinen, V. (2013) J. Biol. Chem., 288, 13467–13480.
59) Satokata, I., Ma, L., Ohshima, H., Bei, M., Woo, I., Nishizawa, K., Maeda, T., Takano, Y., Uchiyama, M., Heaney, S., Peters, H., Tang, Z., Maxson, R., & Maas, R. (2000) Nat. Genet., 24, 391–395.
60) Dudas, M., Sridurongrit, S., Nagy, A., Okazaki, K., & Kaartinen, V. (2004) Mech. Dev., 121, 173–182.
61) Nomura-Kitabayashi, A., Phoon, C.K., Kishigami, S., Rosenthal, J., Yamauchi, Y., Abe, K., Yamamura, K., Samtani, R., Lo, C.W., & Mishina, Y. (2009) Am. J. Physiol. Heart Circ. Physiol., 297, H1617–H1628.
62) Stottmann, R.W., Choi, M., Mishina, Y., Meyers, E.N., & Klingensmith, J. (2004) Development, 131, 2205–2218.
63) Morikawa, Y., Zehir, A., Maska, E., Deng, C., Schneider, M.D., Mishina, Y., & Cserjesi, P. (2009) Development, 136, 3575–3584.
64) Li, L., Lin, M., Wang, Y., Cserjesi, P., Chen, Z., & Chen, Y. (2011) Dev. Biol., 349, 451–461.
65) Mishina, Y. & Snider, T.N. (2014) Exp. Cell Res., 325, 138–147.
66) Noden, D.M. & Trainor, P.A. (2005) J. Anat., 207, 575–601.
67) Jiang, X., Iseki, S., Maxson, R.E., Sucov, H.M., & Morriss-Kay, G.M. (2002) Dev. Biol., 241, 106–116.
68) Sahar, D.E., Longaker, M.T., & Quarto, N. (2005) Dev. Biol., 280, 344–361.
69) Snider, T.N. & Mishina, Y. (2014) Birth Defects Res. C Embryo Today, 102, 324–332.
70) Morriss-Kay, G.M. & Wilkie, A.O. (2005) J. Anat., 207, 637–653.
71) Qiao, T., Wang, G., Xiong, J., Luo, W., & Chen, J. (2015) J. Pediatr. Ophthalmol. Strabismus, 52, 299–304.
72) Levi, B., Kasten, S.J., & Buchman, S.R. (2010) J. Craniofac. Surg., 21, 491–494.
73) Wilkie, A.O., Byren, J.C., Hurst, J.A., Jayamohan, J., Johnson, D., Knight, S.J., Lester, T., Richards, P.G., Twigg, S.R., & Wall, S.A. (2010) Pediatrics, 126, e391–e400.
74) Twigg, S.R. & Wilkie, A.O. (2015) Am. J. Hum. Genet., 97, 359–377.
75) Jabs, E.W., Muller, U., Li, X., Ma, L., Luo, W., Haworth, I.S., Klisak, I., Sparkes, R., Warman, M.L., Mulliken, J.B., Snead, M.L., & Maxson, R. (1993) Cell, 75, 443–450.
76) Liu, Y.H., Tang, Z., Kundu, R.K., Wu, L., Luo, W., Zhu, D., Sangiorgi, F., Snead, M.L., & Maxson, R.E. Jr. (1999) Dev. Biol., 205, 260–274.
77) Warren, S.M., Brunet, L.J., Harland, R.M., Economides, A.N., & Longaker, M.T. (2003) Nature, 422, 625–629.
78) Komatsu, Y., Yu, P.B., Kamiya, N., Pan, H., Fukuda, T., Scott, G.J., Ray, M.K., Yamamura, K., & Mishina, Y. (2013) J. Bone Miner. Res., 28, 1422–1433.
79) Hayano, S., Komatsu, Y., Pan, H., & Mishina, Y. (2015) Development, 142, 1357–1367.
80) Zhao, H., Feng, J., Ho, T.V., Grimes, W., Urata, M., & Chai, Y. (2015) Nat. Cell Biol., 17, 386–396.
81) Zhao, H. & Chai, Y. (2015) J. Dent. Res., 94, 1495–1501.
82) Fukuda, T., Scott, G., Komatsu, Y., Araya, R., Kawano, M., Ray, M.K., Yamada, M., & Mishina, Y. (2006) Genesis, 44, 159–167.
83) Justice, C.M., Yagnik, G., Kim, Y., Peter, I., Jabs, E.W., Erazo, M., Ye, X., Ainehsazan, E., Shi, L., Cunningham, M.L., Kimonis, V., Roscioli, T., Wall, S.A., Wilkie, A.O., Stoler, J., Richtsmeier, J.T., Heuze, Y., Sanchez-Lara, P.A., Buckley, M.F., Druschel, C.M., Mills, J.L., Caggana, M., Romitti, P.A., Kay, D.M., Senders, C., Taub, P.J., Klein, O.D., Boggan, J., Zwienenberg-Lee, M., Naydenov, C., Kim, J., Wilson, A.F., & Boyadjiev, S.A. (2012) Nat. Genet., 44, 1360–1364.
84) Timberlake, A.T., Choi, J., Zaidi, S., Lu, Q., Nelson-Williams, C., Brooks, E.D., Bilguvar, K., Tikhonova, I., Mane, S., Yang, J.F., Sawh-Martinez, R., Persing, S., Zellner, E.G., Loring, E., Chuang, C., Galm, A., Hashim, P.W., Steinbacher, D.M., DiLuna, M.L., Duncan, C.C., Pelphrey, K.A., Zhao, H., Persing, J.A., & Lifton, R.P. (2016) eLife, 5, e20125.
85) Estrada, K.D., Retting, K.N., Chin, A.M., & Lyons, K.M. (2011) J. Bone Miner. Res., 26, 2498–2510.
86) Komatsu, Y. & Mishina, Y. (2016) eLife, 5, e21162.
87) Yin, L., Du, X., Li, C., Xu, X., Chen, Z., Su, N., Zhao, L., Qi, H., Li, F., Xue, J., Yang, J., Jin, M., Deng, C., & Chen, L. (2008) Bone, 42, 631–643.
88) Maruyama, T., Mirando, A.J., Deng, C.X., & Hsu, W. (2010) Sci. Signal., 3, ra40.
89) Lo, K.W., Ulery, B.D., Ashe, K.M., & Laurencin, C.T. (2012) Adv. Drug Deliv. Rev., 64, 1277–1291.
90) Fassbender, M., Minkwitz, S., Strobel, C., Schmidmaier, G., & Wildemann, B. (2014) Int. J. Mol. Sci., 15, 8539–8552.
91) Ghodadra, N. & Singh, K. (2008) Biologics, 2, 345–354.
92) Kim, H.K., Oxendine, I., & Kamiya, N. (2013) Bone, 54, 141–150.
93) Wu, D.T., Bitzer, M., Ju, W., Mundel, P., & Bottinger, E.P. (2005) J. Am. Soc. Nephrol., 16, 3211–3221.
94) Gautschi, O.P., Frey, S.P., & Zellweger, R. (2007) ANZ J. Surg., 77, 626–631.
95) Woo, E.J. (2012) J. Oral Maxillofac. Surg., 70, 765–767.
96) Allendorph, G.P., Read, J.D., Kawakami, Y., Kelber, J.A., Isaacs, M.J., & Choe, S. (2011) PLoS One, 6, e26402.
97) Yoon, B.H., Esquivies, L., Ahn, C., Gray, P.C., Ye, S.K., Kwiatkowski, W., & Choe, S. (2014) J. Bone Miner. Res., 29, 1950–1959.
98) Kamiya, N. & Mishina, Y. (2011) Biofactors, 37, 75–82.
99) Bandyopadhyay, A., Tsuji, K., Cox, K., Harfe, B.D., Rosen, V., & Tabin, C.J. (2006) PLoS Genet., 2, e216.
100) Tsuji, K., Bandyopadhyay, A., Harfe, B.D., Cox, K., Kakar, S., Gerstenfeld, L., Einhorn, T., Tabin, C.J., & Rosen, V. (2006) Nat. Genet., 38, 1424–1429.
101) Okamoto, M., Murai, J., Yoshikawa, H., & Tsumaki, N. (2006) J. Bone Miner. Res., 21, 1022–1033.
102) Mishina, Y., Starbuck, M.W., Gentile, M.A., Fukuda, T., Kasparcova, V., Seedor, J.G., Hanks, M.C., Amling, M., Pinero, G.J., Harada, S., & Behringer, R.R. (2004) J. Biol. Chem., 279, 27560–27566.
103) Tan, X., Weng, T., Zhang, J., Wang, J., Li, W., Wan, H., Lan, Y., Cheng, X., Hou, N., Liu, H., Ding, J., Lin, F., Yang, R., Gao, X., Chen, D., & Yang, X. (2007) J. Cell Sci., 120, 2162–2170.
104) Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W.G., Ross, J., Haug, J., Johnson, T., Feng, J.Q., Harris, S., Wiedemann, L.M., Mishina, Y., & Li, L. (2003) Nature, 425, 836–841.
105) Kamiya, N., Ye, L., Kobayashi, T., Lucas, D.J., Mochida, Y., Yamauchi, M., Kronenberg, H.M., Feng, J.Q., & Mishina, Y. (2008) J. Bone Miner. Res., 23, 2007–2017.
106) Kamiya, N., Ye, L., Kobayashi, T., Mochida, Y., Yamauchi, M., Kronenberg, H.M., Feng, J.Q., & Mishina, Y. (2008) Development, 135, 3801–3811.
107) Kamiya, N., Kobayashi, T., Mochida, Y., Yu, P.B., Yamauchi, M., Kronenberg, H.M., & Mishina, Y. (2010) J. Bone Miner. Res., 25, 200–210.
108) Okamoto, M., Murai, J., Imai, Y., Ikegami, D., Kamiya, N., Kato, S., Mishina, Y., Yoshikawa, H., & Tsumaki, N. (2011) J. Bone Miner. Res., 26, 2511–2522.
109) Shi, C., Zhang, H., Louie, K., Mishina, Y., & Sun, H. (2017) J. Cell. Biochem., 118, 605–614.
110) Kamiya, N., Shuxian, L., Yamaguchi, R., Phipps, M., Aruwajoye, O., Adapala, N.S., Yuan, H., Kim, H.K., & Feng, J.Q. (2016) Bone, 91, 53–63.
111) Lim, J., Shi, Y., Karner, C.M., Lee, S.Y., Lee, W.C., He, G., & Long, F. (2016) Development, 143, 339–347.
112) Zhang, Y., McNerny, E.G., Terajima, M., Raghavan, M., Romanowicz, G., Zhang, Z., Zhang, H., Kamiya, N., Tantillo, M., Zhu, P., Scott, G.J., Ray, M.K., Lynch, M., Ma, P.X., Morris, M.D., Yamauchi, M., Kohn, D.H., & Mishina, Y. (2016) Bone, 88, 74–84.
113) Ahrengart, L. (1991) Clin. Orthop. Relat. Res., 49–58.
114) Bal, B.S., Lowe, J.A., E Gietler, A., & Aleto, T.J. (2010) J. Arthroplasty, 25, 538–540.
115) Chen, H.C., Yang, J.Y., Chuang, S.S., Huang, C.Y., & Yang, S.Y. (2009) Burns, 35, 857–862.
116) Kraft, C.T., Agarwal, S., Ranganathan, K., Wong, V.W., Loder, S., Li, J., Delano, M.J., & Levi, B. (2016) J. Trauma Acute Care Surg., 80, 156–165.
117) Potter, B.K., Burns, T.C., Lacap, A.P., Granville, R.R., & Gajewski, D. (2006) J. Am. Acad. Orthop. Surg., 14(Supplement), S191–S197.
118) Lee, E.K., Namdari, S., Hosalkar, H.S., Keenan, M.A., & Baldwin, K.D. (2013) J. Shoulder Elbow Surg., 22, 716–722.
119) Peterson, J.R., De La Rosa, S., Eboda, O., Cilwa, K.E., Agarwal, S., Buchman, S.R., Cederna, P.S., Xi, C., Morris, M.D., Herndon, D.N., Xiao, W., Tompkins, R.G., Krebsbach, P.H., Wang, S.C., & Levi, B. (2014) Sci. Transl. Med., 6, 255ra132.
120) Agarwal, S., Loder, S., Brownley, C., Cholok, D., Mangiavini, L., Li, J., Breuler, C., Sung, H.H., Li, S., Ranganathan, K., Peterson, J., Tompkins, R., Herndon, D., Xiao, W., Jumlongras, D., Olsen, B.R., Davis, T.A., Mishina, Y., Schipani, E., & Levi, B. (2016) Proc. Natl. Acad. Sci. USA, 113, E338–E347.
121) Forsberg, J.A., Davis, T.A., Elster, E.A., & Gimble, J.M. (2014) Sci. Transl. Med., 6, 255fs237.
122) Agarwal, S., Drake, J., Qureshi, A.T., Loder, S., Li, S., Shigemori, K., Peterson, J., Cholok, D., Forsberg, J.A., Mishina, Y., Davis, T.A., & Levi, B. (2016) PLoS ONE, 11, e0156253.
123) Kan, L., Hu, M., Gomes, W.A., & Kessler, J.A. (2004) Am. J. Pathol., 165, 1107–1115.
124) Kan, L., Kitterman, J.A., Procissi, D., Chakkalakal, S., Peng, C.Y., McGuire, T.L., Goldsby, R.E., Pignolo, R.J., Shore, E.M., Kaplan, F.S., & Kessler, J.A. (2012) J. Neurol., 259, 2644–2655.
125) Kan, L., Lounev, V.Y., Pignolo, R.J., Duan, L., Liu, Y., Stock, S.R., McGuire, T.L., Lu, B., Gerard, N.P., Shore, E.M., Kaplan, F.S., & Kessler, J.A. (2011) J. Cell. Biochem., 112, 2759–2772.
126) Salisbury, E., Rodenberg, E., Sonnet, C., Hipp, J., Gannon, F.H., Vadakkan, T.J., Dickinson, M.E., Olmsted-Davis, E.A., & Davis, A.R. (2011) J. Cell. Biochem., 112, 2748–2758.
127) Yu, P.B., Deng, D.Y., Lai, C.S., Hong, C.C., Cuny, G.D., Bouxsein, M.L., Hong, D.W., McManus, P.M., Katagiri, T., Sachidanandan, C., Kamiya, N., Fukuda, T., Mishina, Y., Peterson, R.T., & Bloch, K.D. (2008) Nat. Med., 14, 1363–1369.
128) Yu, P.B., Hong, C.C., Sachidanandan, C., Babitt, J.L., Deng, D.Y., Hoyng, S.A., Lin, H.Y., Bloch, K.D., & Peterson, R.T. (2008) Nat. Chem. Biol., 4, 33–41.
129) Shimono, K., Tung, W.E., Macolino, C., Chi, A.H., Didizian, J.H., Mundy, C., Chandraratna, R.A., Mishina, Y., Enomoto-Iwamoto, M., Pacifici, M., & Iwamoto, M. (2011) Nat. Med., 17, 454–460.
130) Wang, H., Lindborg, C., Lounev, V., Kim, J.H., McCarrick-Walmsley, R., Xu, M., Mangiavini, L., Groppe, J.C., Shore, E.M., Schipani, E., Kaplan, F.S., & Pignolo, R.J. (2016) J. Bone Miner. Res., 31, 1652–1665.
131) Kaplan, F.S., Xu, M., Seemann, P., Connor, J.M., Glaser, D.L., Carroll, L., Delai, P., Fastnacht-Urban, E., Forman, S.J., Gillessen-Kaesbach, G., Hoover-Fong, J., Koster, B., Pauli, R.M., Reardon, W., Zaidi, S.A., Zasloff, M., Morhart, R., Mundlos, S., Groppe, J., & Shore, E.M. (2009) Hum. Mutat., 30, 379–390.
132) Pignolo, R.J., Bedford-Gay, C., Liljesthrom, M., Durbin-Johnson, B.P., Shore, E.M., Rocke, D.M., & Kaplan, F.S. (2016) J. Bone Miner. Res., 31, 650–656.
133) Shore, E.M., Xu, M., Feldman, G.J., Fenstermacher, D.A., Cho, T.J., Choi, I.H., Connor, J.M., Delai, P., Glaser, D.L., LeMerrer, M., Morhart, R., Rogers, J.G., Smith, R., Triffitt, J.T., Urtizberea, J.A., Zasloff, M., Brown, M.A., & Kaplan, F.S. (2006) Nat. Genet., 38, 525–527.
134) Nakahara, Y., Katagiri, T., Ogata, N., & Haga, N. (2014) Am. J. Med. Genet. A., 164A, 220–224.
135) Nakahara, Y., Suzuki, R., Katagiri, T., Toguchida, J., & Haga, N. (2015) Hum. Genome Var., 2, 15055.
136) Chakkalakal, S.A., Zhang, D., Culbert, A.L., Convente, M.R., Caron, R.J., Wright, A.C., Maidment, A.D., Kaplan, F.S., & Shore, E.M. (2012) J. Bone Miner. Res., 27, 1746–1756.
137) Hatsell, S.J., Idone, V., Wolken, D.M., Huang, L., Kim, H.J., Wang, L., Wen, X., Nannuru, K.C., Jimenez, J., Xie, L., Das, N., Makhoul, G., Chernomorsky, R., D’Ambrosio, D., Corpina, R.A., Schoenherr, C.J., Feeley, K., Yu, P.B., Yancopoulos, G.D., Murphy, A.J., & Economides, A.N. (2015) Sci. Transl. Med., 7, 303ra137.
138) Hino, K., Ikeya, M., Horigome, K., Matsumoto, Y., Ebise, H., Nishio, M., Sekiguchi, K., Shibata, M., Nagata, S., Matsuda, S., & Toguchida, J. (2015) Proc. Natl. Acad. Sci. USA, 112, 15438–15443.
139) Chakkalakal, S.A., Uchibe, K., Convente, M.R., Zhang, D., Economides, A.N., Kaplan, F.S., Pacifici, M., Iwamoto, M., & Shore, E.M. (2016) J. Bone Miner. Res., 31, 1666–1675.
140) Katagiri, T. (2016) Trends Biochem. Sci., 41, 119–121.
著者紹介Author Profile
 三品 裕司(みしな ゆうじ)
三品 裕司(みしな ゆうじ)ミシガン大学歯学部生命材料学科マウス発生遺伝学研究室教授.薬学博士.
略歴1958年東京に生まれる.86年東京大学大学院薬学系研究科修了,92年テキサス大学MDアンダーソンがんセンター博士研究員,98年米国立衛生科学研究所環境衛生科学研究所生殖発生毒性学部門分子発生学研究室室長,2008年ミシガン大学歯学部生命材料学科マウス発生遺伝学研究室准教授,14年より現職.
研究テーマと抱負生き物の形はどうきまるのかという疑問を幼少のころにもち,その解答を発生学に求めて現在に至る.増殖因子の下流に位置する遺伝子ネットワークに注目してなぜ子供は親の顔に似るのかを解き明かしたい.
ウェブサイトhttp://www.dent.umich.edu/about-school/department/bms/mishina-lab