一酸化窒素(nitric oxide, NO)が血管内皮細胞から放出される平滑筋弛緩因子として同定されたのは1980年代である.ほぼ時を同じくして,NOが環状グアノシン3′,5′-一リン酸(環状GMP, cGMP)の産生を増大すること,加えてNOがアルギニンから合成されることが見いだされた.その後,ガス状分子であるNOをシグナル分子とする新規な情報伝達系が,血圧制御,血小板凝集阻害等のさまざまな生理作用を引き起こすことが明らかにされている.NOの受容体は,可溶性のヘム酵素であるグアニル酸シクラーゼであり,そのヘム鉄にNOが結合すると環状GMPの産生は著しく活性化される.本酵素は,NOの感知に特化しており,他のヘムタンパク質にはみられない特異な性質を示す.
本稿では,可溶性グアニル酸シクラーゼの構造と機能,特にヘム鉄の配位子に対する選択性を制御する因子に焦点を当て,我々が近年見いだした知見も交えて概説する.
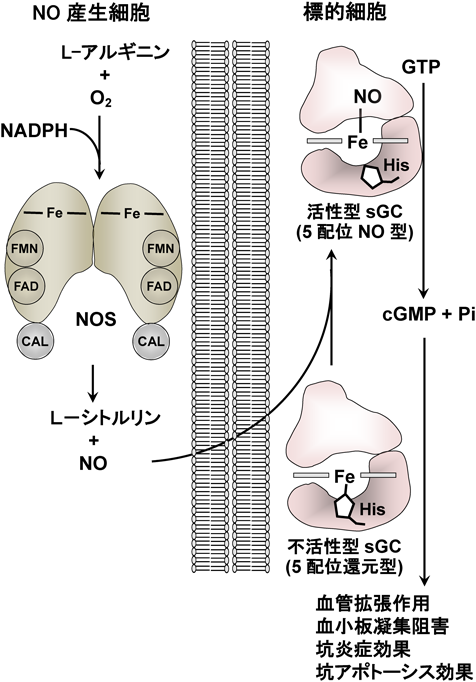
真核生物では,NOはアルギニンから合成される.この反応は,シトクロムP450に分類されるヘム酵素であるNO合成酵素(NOS)により触媒される酸素添加反応であり,NO中の酸素原子は酸素分子に由来する(図1).血管内皮細胞などのNO産生細胞で合成されたNOは,チャネルを介さずに産生細胞の細胞膜を迅速に透過して標的細胞に到達し,標的細胞内に存在するNO受容体である可溶性グアニル酸シクラーゼ(soluble guanylate cyclase:sGC)に結合してcGMPの産生を活性化する.NOは不対電子を持つラジカル種であり,酸素分子と反応するのでその寿命は短いが,ヘム酵素であるsGCにはNOを効率よく捕捉する仕組みが備わっている.図1に示すように,不活性な還元型sGC(5配位還元型)では,ヘムの第五配位座にヒスチジン残基(His)が配位しており,その逆側の第六配位座は空位である.こののち,第五配位座側をproximal side,第六配位座側をdistal sideと表記する.NOは空位の第六配位座に結合して6配位NO型を生成するが,それは不活性である.6配位NO型は速やかに5配位NO型に変換され,その配位構造変化に伴いグアノシン三リン酸(GTP)をcGMPに変換する活性が約200程度増加する(図1,詳細は後述).NO存在下におけるcGMP産生活性は,ヘムあたりの回転数で表すと2000/分(37°C)以上に達する.産生されたcGMPは,cGMP依存性タンパク質リン酸化酵素等を介して,血管拡張,血小板凝集阻害等に関与している.
sGCがヘテロ二量体であることが証明されたのは1986年であり1),現在では,その二量体を構成するサブユニットはα1, β1と表記されている.引き続き,それらとは異なるα, βサブユニットが遺伝子解析から見いだされ,それぞれα2, β2と命名された.α1鎖とβ1鎖はほとんどの組織で発現しているので,生理的に重要な分子種はα1β1ヘテロ二量体と考えられ,後述するsGCの酵素学的解析はこの分子種を用いて行われている.
β2鎖は主に腎臓で発現しているが,単離されてはいない2).β2鎖はβ1鎖と異なり,イソプレノイド化される配列を含んでおり膜結合性と予想されるが,その局在はいまだに不明である.
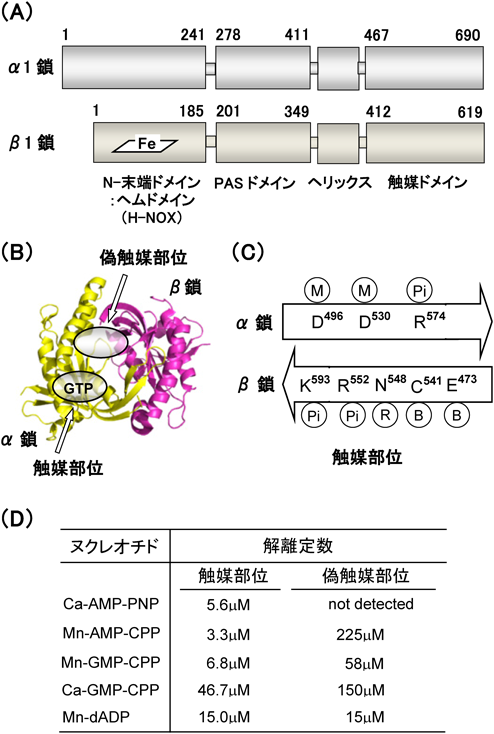
ラットsGCのα1鎖は690残基のアミノ酸,β1鎖は619残基のアミノ酸から構成されている(図2A).ラットのα1とβ1鎖のアミノ酸の相同性は約30%であり,ヒト,ウシでもほぼ同様である.真核生物種間におけるα1鎖の相同性,β1鎖の相同性はきわめて高く,約80%に及ぶ.特に,α1ならびにβ1鎖のC末端側の配列における相同性はきわめて高度に保存されていること,加えてこの領域の配列がアデニル酸シクラーゼの触媒ドメインとの相同性が高いことから,これらの領域が触媒部位を構成しているものと考えられた.
α1ならびにβ1鎖は相互に類似したドメイン構造を持ち,それぞれ四つのドメイン,すなわちN末端ドメイン(β鎖ではヘムドメインと呼称),PAS(Per/Arnt/Sim)ドメイン,ヘリックス部位(coiled-coil),触媒ドメインから構成されている(図2A).これまで多くの研究グループが,組織から抽出したsGCあるいはcDNAから発現した完全長sGCの結晶化を試みたものと思われるが,いずれも不成功に終わっている.しかし,sGCを構成するドメインのうち,ヘムドメイン以外のPASドメイン,ヘリックス部位,触媒ドメインの結晶構造は,対応する部位の発現タンパク質を用いて明らかにされている.以下に,筆者の興味の対象であるヘムドメインと触媒ドメインについて以下に詳述する.
1990年代にアデニル酸シクラーゼ(AC)の2種類の触媒ドメインが二量体を形成して触媒部位を形成すること,ならびにその二量体の結晶構造が解明された3).その結果,触媒部位は,二つのドメインが逆方向(head to tail)に会合した接触面(interface)に形成されることが明らかになった.ACの触媒ドメインのアミノ酸配列がsGCのC末端領域のそれと相同性が高く,ACの結晶構造を鋳型にしたモデルが構築された3).以下に解説するように,このモデルの妥当性は,ヒトsGCのα1触媒ドメインとβ1触媒ドメイン複合体の結晶構造から裏づけられている4, 5)(図2B).
sGCの触媒ドメイン複合体の結晶構造とそれに対応するACの結晶構造との相同性ならびにアミノ酸変異体の解析から,sGCの触媒機能を担うアミノ酸残基が同定されている4, 5).具体的には,図2Cに示すように,①αD496とαD530はMg2+を結合する部位であり,GTPの3′-OHとαリン酸の活性化に,②βE473とβC541は塩基の認識に,③αR574, βR552とβK593はリン酸基との相互作用に関わっている.ACとsGCでは塩基を認識する残基が異なるので,sGCの塩基認識に関わるβE473とβC541を,ACにおける塩基認識残基に置換(βE473K, βC541D)すると,このsGC変異体はNOの結合により活性化されるアデニル酸シクラーゼ活性を示す6).
アデニル酸シクラーゼの結晶構造解析は,触媒部位とそれに類似の部位(偽触媒部位,pseudocatalytic site)の存在を明らかにするとともに,後者の偽触媒部位は活性化剤であるフォルスコリンの結合部位であることを明らかにした3).sGCのαとβ鎖のC末端触媒ドメインからなるヘテロ二量体の結晶構造においても,ACと類似の偽触媒部位が見いだされている4)(図2B).sGCでは,触媒部位を構成するαD496残基が,偽触媒部位ではGly(βG426)に置き換わっているためsGC活性が欠損しているものと考えられている.
上記のsGCのC末端触媒ドメインの結晶構造が報告される以前に,我々はα1β1ヘテロ二量体sGCとヌクレオチドとの結合親和性を解析し,予想外なことにある種のヌクレオチドがsGCの1分子に2分子結合する結果を得ていた7).その結果をまとめると①基質GTPに対して拮抗阻害剤として作用するATP誘導体,adenosine 5′-(β,γ-imido)triphosphate(AMP-PNP)が,意外なことに高い親和性で1分子結合すること,②ATP誘導体であるα,β-methylene adenosine 5′-triphosphate(AMP-CPP)あるいはGTP誘導体であるα,β-methylene guanosine 5′-triphosphate(GMP-CPP)は,低親和性と高親和性部位のそれぞれに1分子結合することがわかった(図2D).また,拮抗阻害剤であるAMP-PNPは,GMP-CPPの高親和性部位への結合を阻害するので,高親和性部位は触媒部位であり,低親和性部位が偽触媒部位であることが推定された.これらの結果は,提案されたモデル,すなわち偽触媒部位がシクラーゼ反応活性を欠いていることと矛盾しない.我々の結果は,NO等の外来性配位子が結合していない還元型sGCとヌクレオチドの結合の解析に基づいたものであり,この場合,触媒部位と偽触媒部位はそれぞれ独立にヌクレオチドを結合する.最近の速度論的解析によれば,NOを結合したsGCでは両部位間にアロステリック相互作用を考慮する必要があるという8).
意外なことに,上記の我々が得た結果は,sGCが示す塩基認識の特異性は従来想定されていたほど厳密ではなく,グアニンのみならずアデニンもほぼ同程度の親和性で両部位に結合することを示している.最近の報告では,ATPはもとよりイノシン5′-三リン酸(ITP)もsGCの基質となり,かなりの高活性でcIMPを生成するという9).また,sGC活性は非拮抗的にATPにより阻害されることから,sGCがATPセンサーとして機能しているとの報告もある10, 11).この場合,ATPは,生理的濃度範囲(数mM程度)で非拮抗阻害剤として作用するので,ATPの結合部位の候補として,低親和性の偽触媒部位が推定された7).このように,偽触媒部位は,ATP等のヌクレオチドが結合すると触媒ドメインと相互作用して活性調節に関わっているらしい.
ウシ肺から精製した酵素は,鉄–ポルフィリン錯体であるヘムに由来する赤褐色を示す.ヘムの配位構造は,経験的ではあるが,光吸収スペクトルの極大値からほぼ正確に決定することができる.
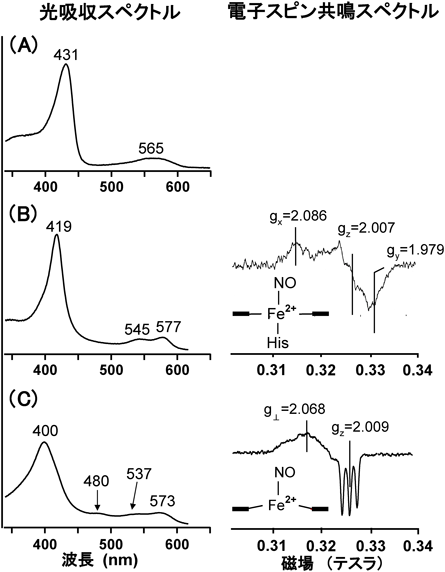
還元型sGC(ヘム鉄が二価,Fe2+)は,酸素が存在するにも関わらず安定であり,図3Aに示すように431 nmにソーレー帯を示す(400~450 nm前後の強い光吸収帯をソーレー帯と呼ぶ).このようなソーレー帯を示す還元型ヘム鉄は,5配位型に帰属される.ヘム鉄は,本来,ポルフィリンの四つのピロール環の窒素原子が配位した4配位型であるので,5配位型とは,以下に記すようにさらにもう一つの分子(内在性配位子)がヘム鉄に配位している状態を意味する.
5配位型である還元型sGC(Fe2+)のヘム鉄の内在性配位子がβ鎖のヒスチジン残基(His105)であることは部位特異的変異法,β鎖の欠損変異体の解析から確認された12, 13).ヘムドメインは,現在では,原核生物から真核生物にひろく高度に保存されたヘム結合ドメインであることが知られており,heme-nitric oxide and oxygen binding domain(H-NOX)と命名されている14).現在まで,多くのH-NOXタンパク質が知られているが,外来性配位子に対する反応性に基づき,以下の2種類に分類される.
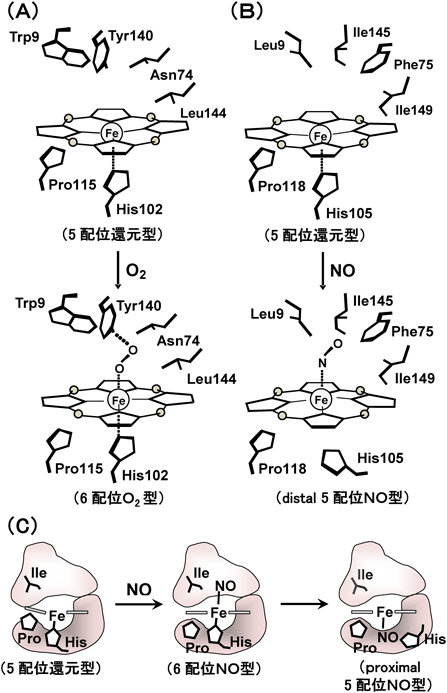
その一つは,酸素を結合する能力を持ち,加えて第六配位座にNOが配位すると安定な6配位NO型を形成するグループである(図4A).他方は,酸素結合能を欠き,NOと反応したとき安定な5配位NO型を形成するグループである(図4B).以下に述べるように,酸素結合能を示すH-NOXでは,第六配位座側のヘムポケットにTyr残基が存在する14, 15).一方,酸素結合能を欠くグループにはそれに対応する位置にLeuあるいはIle残基が存在する15, 16).
Thermoanaerobactor tengcongensis由来の酸素結合性H-NOXドメイン(Tt H-NOX)のヘム周辺の構造を,図4Aに模式的に示した.他のH-NOXドメインホモログにおいても,ヘムの第五配位座には例外なくHis残基が配位している.Tt H-NOXの第六配位座側のヘムポケットを構成する代表的なアミノ酸残基は,Trp9, Tyr140, Leu144等の疎水性残基である.これらの残基のうち,Tyr残基は,他の酸素結合性H-NOXドメインにおいても例外なく対応する位置に保存されている15).また,H-NOXドメインの酸素結合能の有無に関わらずTrp9やLeu144の位置にLeu, IleあるいはPhe等の疎水性残基が存在する.興味深いことに,proximal sideには,酸素結合能の有無に関わらずPro残基がヘムと接触する位置に高度に保存されている.後述するように,この接触によりヘムは歪んでいる(図4C)15).sGCにおいても対応する位置にPro残基が存在する.
sGCを含めて酸素結合能を欠くH-NOXでは,上記のTyr140に対応する残基がLeuあるいはIle等の疎水性アミノ酸に置換されている.Tt H-NOXホモログの構造を鋳型にした相同性モデル解析から,sGCにおいてもdistal sideのヘムポケットを形成するアミノ酸のほとんどが疎水性のアミノ酸であることが示唆されている(図4B).繰り返すが,sGCの場合,安定に存在するNO複合体は5配位型であり,生理的条件下では安定な酸素結合型は形成されない.このように,sGCでは,distal sideのTyr残基がIleに置換されており,これが酸素結合能を欠く要因と推定されている.これが酸素存在下においても選択的にNO感知することを可能にする構造的要因の一つである(後述).
6. NOの結合に伴うヘム鉄の配位構造変化とその機構
1992年,TraylorとSharmaは,「Why NO?」という題名で,NOの結合に伴うヘムの配位構造変化に基づいた分子内シグナル伝達機構を予測した17).その内容は,Hisが配位したヘム鉄(Fe–His)にNOが結合すると,NOがFeを自身の方に引き寄せ(negative trans効果),その結果FeとHis残基間の結合が切断された5配位NO型が形成され,sGCが活性化されるというものである.NOのnegative trans効果は,すでにヘモグロビンあるいはモデルヘム錯体において知られていたので,特に目新しいものではなかったが,当時は信頼にたるsGCの光吸収スペクトルすら報告がなく,そのモデルの妥当性が実証されたのは数年後のことである18).
1990年代の半ばに,筆者らは電気泳動的に均一なsGC標品を得ることができるようになり,NOとの反応をストップド・フローにより解析した19).この手法は,2種類の溶液を迅速に混合し,混合後約2ミリ秒後からの反応過程を追跡できる混合装置を備えた分光光度計を用いる.
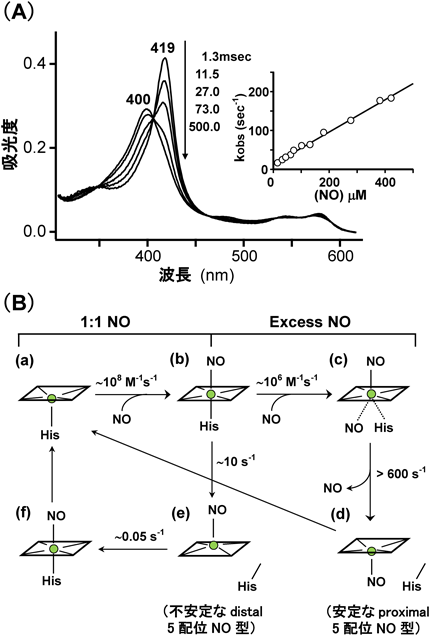
sGCとNOとの反応は非常に速く,両者の混合後,装置のdead time(数ミリ秒)以内に還元型sGCは6配位NO型と予想される419 nmに吸収極大を示す分子種に変換し,引き続いて1秒以内に5配位NO型と予想される400 nm分子種へと等吸収点を通って変換する結果が得られた(図5A).しかし,他のヘムタンパク質,たとえばミオグロビンとは異なり,sGCでは還元型から6配位NO型への生成過程が観測できない.これは,sGCにおける6配位NO型の生成過程がきわめて速く,装置のdead time内で終了するためである.
419 nm分子種が6配位NO型であると予想できたが,最終的に,それは電子スピン共鳴(ESR)法により確認された19).この方法では,ヘムに配位したNOの不対電子に由来するESRシグナルから,6配位NO型と5配位NO型を相互に識別できる.400 nm分子種は3本の鋭いESRシグナルを示すので,5配位NO型であることが確認された(図3C).一方,419 nm分子種のESRシグナルは5配位NO型とは明瞭に異なり,6配位NO型であることが確認できた(図3B).したがって,sGCでは,いったん6配位NO型が生成した後,6配位NO型のヘム鉄とHis残基間の結合が切断されて5配位NO型に変換することがわかった.反応は以下のようにまとめられる. 6配位NO型から5配位NO型に迅速に変換するヘムタンパク質の例はまれであり,通常は6配位NO型の状態にとどまる.したがって,容易に5配位NO型の生成が観察されるsGCでは,他のヘムタンパク質とは異なりFe–His間の結合が弱いことが予想された.Fe–His間の結合の強さは,Fe–His間の伸縮振動(νFe–His)から評価でき,それは共鳴ラマン法で測定可能である.sGCは204 cm−1にνFe–Hisに帰属される共鳴ラマン帯を示し,その値はミオグロビンのそれより有意に低く20),これまで報告されているヘムタンパク質中では最も低波数に位置する.先述したTt H-NOXドメインのνFe–Hisは217 cm−1であり,その6配位NO型は安定であり容易に5配位NO型に変換しない.これらの結果は,予想どおりsGCのFe–His結合が他のヘムタンパク質より異常に弱く,そのためFe–His間の結合が容易に切断されて5配位NO型が生成するものと結論されている.このように,sGCは,1ミリ秒以下で終わる速い反応速度(>108 M−1s−1)でNOを捕捉し(6配位NO型の形成),さらに安定な5配位NO型に迅速に変換される.
しかしこれで一件落着とはならなかった.我々の論文発表後に,Zhaoらは5配位NO型の生成速度がNOに依存することを報告したからである21)(たとえば図5A,挿入図).この結果は,6配位NO型から5配位NO型の生成には,さらにもう1分子のNOが関与することを意味するものであった.我々もNO濃度依存性を検討してはいたが,当時使用していた装置では酸素との反応性が高いNOを定量的に扱うことが困難であり,そのためこれまで報告のない新規な反応を見逃した.
ほぼ時を同じくして,シトクロムc′においても,sGCの場合とほぼ同様な機構で6配位NO型から5配位NO型への変換が明らかにされ,その安定な5配位NO型の結晶構造が報告された22).予想外なことに,NOは空位である第六配座側(distal side)ではなく,もともとHis残基が結合していたヘムの第五配座側(proximal side)に結合していたのである.sGCにおける5配位NO型の生成機構は完全に解明されたとはいいがたいが,Martinらの反応速度論的解析から得られた結果は,シトクロムc′と類似の5配位NO型形成機構を支持するものである23).図5Bに示した2種類の5配位型NO型(分子種dとe)に注目していただきたい.6配位NO型(b)からproximal 5配位型NO型(d)への変換過程でヘムの両側にNOを配位した分子種(c)(dinitrosyl heme)の生成が予想されている.cからdへの変換速度は,>600 s−1ときわめて速く(半減期1ミリ秒以下),この短寿命分子種cを捕捉することは現在の混合法による手段では不可能であるが,モデルヘム錯体ではcに相当する分子種が低温(−70°C)で捕捉されている.これらの解析結果から,sGCのNOに対する親和性(解離定数:Kd)は,10−9 M以下と見積もられる.
Shewanella oneidensis由来のH-NOXヘムドメインは,sGCの場合と同様に酸素結合能を欠失したヘムドメインであり,第六配位座側にTyr残基を欠く.その結晶構造が最近報告され,過剰のNO存在下で生成された5配位NO型では,シトクロムc′の場合と同様に,proximal 5配位NO型の生成が確認されている16)(図4C).驚くべきことに,第五配位座のHis残基は5配位NO型形成に伴い,8.5 Åもヘム鉄から移動する.これに伴い,Pro残基との接触により歪んでいたヘムの歪みが解消され,ヘムはほぼ平面状になる.このようなヘムの構造変化が,NOによる本酵素の活性化のトリガーであるとの説が提案されている.しかし,このようなNO結合に伴うヘム面の歪みの解消は,安定な6配位NO型を生成するTt H-NOXでも観察されているので,活性化の主要な要因とは考えにくい.したがって,Fe–His結合の切断により引き起こされる構造変化が活性化の主な要因と思われる.いずれにしても,過剰なNO存在下におけるsGCの活性化は,proximal sideにNOが配位した5配位型の生成に伴うHis残基の移動によるものと考えてよさそうである.
細菌由来のH-NOXヘムドメインの構造に基づいて,sGCの5配位NO型生成機構を議論することに異論がないわけではない.Yooらは,ピコ秒からミリ秒の広い時間範囲にわたるレーザーフラッシュ時間分解分光法によりsGCのNO結合過程を解析している24).この方法では,光解離後の分子種が元の5配位NO型に戻るNOの再結合過程を,ピコ秒からミリ秒の広い時間範囲で追跡できる.図5Bの分子種c(dinitrosyl heme)が6配位NO型である分子種bから形成されるとすると,この場合,proximal sideに結合する2個目のNOは溶液からタンパク質内部への拡散により供給されなければならない.このタンパク質内部への拡散速度はマイクロ秒~ミリ秒の範囲と見積もられるが,この時間範囲において,dinitrosyl hemeに対応する分子種は検出されていないので,proximal 5配位NO型の生成を疑問視している.
先述したように,還元型sGCは,その第六配位座が空位であるにも関わらず酸素存在下においても酸素化型(Fe2+–O2)を生成しない.つまり,sGCは,酸素存在下の生体内環境において,低濃度のNOを感知することが可能である.NOの高感度感知に合目的なこのような性質は,これまで他のヘムタンパク質では報告されていない特異なものである.たとえばヘモグロビンでは,その還元型ヘム鉄は容易に酸素を結合して,酸素化型ヘモグロビンを生成する.
先述したように,酸素化型を安定に生成するH-NOXでは,ヘムポケット内のTyr残基の水酸基と結合酸素間の水素結合形成が,結晶構造解析から確認されている15)(図4A).また,このTyr残基をLeu残基に置換すると,その変異体の酸素親和性は著しく減少するので,Boonらは,sGCが酸素に対する反応性を欠く要因として,水素結合の重要性を指摘した14).
Martinらはこれを受けて,ヘムポケット内のIle145(図4B参照)をTyrに変換した完全長の変異sGCを用いて酸素との反応性を検討した25).予想に反しこの変異体は酸素化型を安定に形成する能力を欠いていた.逆に,6配位型NOは安定化される.したがって,distal sideのTyr残基はNOと水素結合を形成して6配位NO型を安定化しているようにもみえる.この結果は多様に解釈されるとは思うが,酸素結合能の欠損には水素結合以外の他の要因が含まれている可能性を示唆していた.
上記の結果が報告される以前に,我々は低温下(−180°C)におけるX線吸収スペクトル法(extended X-ray absorption fine structure:EXAFS)によりsGCのヘム鉄配位構造の解析に着手していた.EXAFSは,ヘム鉄に配位している原子間の結合距離の解析に適した手段である.意外なことに,低温(−180°C)では嫌気条件下と好気条件下において還元型酵素は異なるEXAFSを示した.嫌気条件下ではヘム鉄は還元型5配位であるが,酸素が存在する好気条件下では6配位型の分子種が生成していたのである26).
EXAFSでは配位している分子種の同定は困難であるので,EXAFSと同じ条件下,すなわち液体窒素温度での光吸収スペクトルから,その分子種を解析した.−180°Cで測定した光吸収スペクトルは,嫌気条件下では室温でのそれと同様に5配位型であるが,酸素存在下では鋭い543 nmと577 nmに吸収極大を示す6配位型に変換した.それらの吸収極大値は酸素化型ヘモグロビンのそれにほぼ一致するので,凍結過程で還元型sGCの酸素親和性が上昇して酸素化型が生成するものと結論された.EXAFSスペクトルの解析から,酸素化型の配位構造は6配位型であることが確認されたので,酸素の結合はFe–His結合を切断しない.このように,sGCは,酸素結合能を欠いているわけではなくその親和性をきわめて低い状態に保ち,NOに対する高い感知機能を維持している26).
このような低酸素親和性が,ヘム鉄の電子密度に依存していることは容易に想像されたので,その尺度であるヘム鉄の酸化還元電位を測定した.sGCの酸化還元電位は約+200 mVであり,これまで測定された5配位高スピン型ヘムタンパク質の中では最も高い26, 27).このような高い電位は還元型ヘム鉄の電子密度が低いことを意味し,酸素に対する低親和性,酸素によるヘム鉄の自動酸化を防いで還元型ヘム鉄の安定化をもたらす.詳細は省くが,先述したdistal sideの水素結合供与残基の欠除に加えて,弱いFe–His結合が酸素に対する低親和性を引き起こす要因の一つであり,sGCの選択的なNO感知機構を支えている.
生体内では,ヘムオキシゲナーゼ(heme oxygenase)の作用により一酸化炭素(CO)が産生されることが知られており,それがsGC/cGMP情報伝達系のシグナル分子として機能するのか否かは生理的に重要な課題である.還元型sGCに対するCOの親和性(解離定数:Kd)は約300 μMであり,その親和性は5配位還元型ヘムタンパク質としては例外的に低い(たとえばミオグロビンでは1 μM以下).加えて,解離速度定数(CO複合体からCOが解離する速度定数)は約15 s−1(半減期:~50ミリ秒)と速く,同程度のNOが存在する条件下ではsGC–CO複合体が生成したとしても速やかにNO複合体に変換される.さらに,sGC–CO複合体におけるcGMP産生能はsGC–NO複合体の数%であることから,生理的条件下でCOがsGC/cGMP情報伝達系においてシグナル分子として作用する可能性は低いと考えられた.
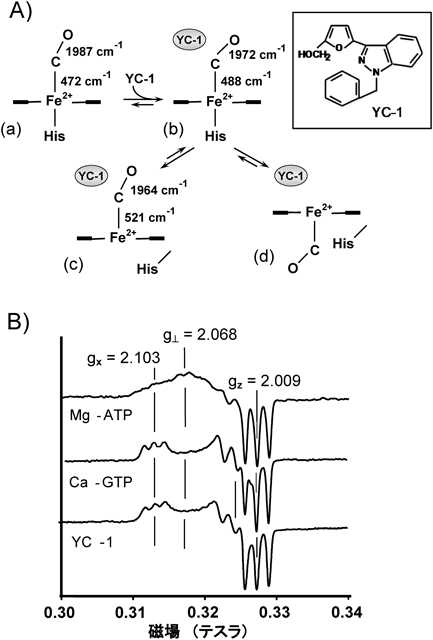
しかしながら,1996年に坑血小板凝集作用を示す薬剤であるYC-1が,sGC–CO複合体の活性をsGC–NOのそれと同程度に活性化することが見いだされ,COシグナル分子説が再び重要視されるに至った28).YC-1結合によるヘムの配位構造変化と活性化との関連は共鳴ラマン法により解析され,Fe–CO伸縮振動(νFe–CO)に帰属されるラマン帯が475 cm−1から488 cm−1へ高波数シフトすることが報告された29)(図6).つまり,488 cm−1のνFe–COを与える分子種は,YC-1が結合した6配位CO型である(図6Aのb).しかし我々は,YC-1結合により生成される分子種はそれだけではなく,別の分子種が形成されることを見いだした30).この分子種は,521 cm−1にνFe–COを,1964 cm−1にC–O伸縮振動(νC–O)を与える.詳細は省くが,この分子種の配位構造は6配位CO型ではなく5配位CO型に帰属されるものである.したがって,YC-1結合により,6配位CO型(図6Aのb)とFe–His残基が切断された5配位CO型(図6Aのcあるいはd)の両者が生成される.その後,YC-1の誘導体であるBAY41-2272においても同様な結果が報告されている31).5配位CO型の生成と活性化との関連についてはいまだに議論のあるところではあるが,我々の知る限り,5配位CO型の生成は,ヘムタンパク質においては初めての例である.なお,5配位CO型の構造としてdistal 5配位CO型(図6Aのc)が推定されるが,proximal 5配位CO型(図6Aのd)の生成も否定できない.これは,上記の条件下で生成する5配位CO型の割合が5~20%程度であること,加えてYC-1あるいはBAYが結合していない6配位CO型が共存するため,5配位CO型の生成機構の速度論的解析が困難なことによる.残念ながら,いまだにYC-1と同等の活性化能を示す天然化合物が報告されていないので,5配位CO型形成機構の解析は生理的に重要であるとは思えないが,COがproximal sideに結合した5配位CO型(図6Aのd)の形成を示唆する最近の速度論的解析結果を以下に紹介する.
2′-deoxy-3′-guanosine monophosphate(2′-d-3′-GMP)はGTP結合部位の近傍に位置するP-siteと呼ばれる部位に結合し,条件に依存するが,非拮抗阻害あるいは不拮抗阻害様式で,シクラーゼ反応を阻害する27).活性化剤の非存在下ではその阻害定数(Ki)は約500 μMであり阻害作用は弱いが,NO, BAY41-2272等の活性化剤が存在するとKiは0.1~10 μMに減少し,強力な阻害作用を示す.
BAY41-2272と2′-d-3′-GMPの両者がsGCに結合すると,そのCO結合型ヘム鉄の約45%が5配位CO型に変換する.つまり,この条件下では,BAY41-2272が結合した6配位CO型と5配位CO型がほぼ等量存在し,それ以外の分子種は存在しない.BAYと2′-d-3′-GMPの共存下でCO結合型の形成をラピッドスキャン法で追跡すると,混合後直ちに423 nmにソーレー帯の吸収極大を示す6配位CO型が生成し,それがゆっくりと418 nmに吸収極大を示す別種のCO型(418 nm CO型)に変換するのが観察される.この418 nm CO型は,6配位CO型と5配位CO型の等量混合物と推定される.奇妙なことに418 nm CO型の生成速度はCO濃度依存性を示す.つまり,6配位CO型から5配位CO型への変換には少なくとももう1分子のCOが関与している.これは先述したproximal 5配位NO型の生成機構に類似しており,この条件下ではproximal sideにCOが結合した5配位CO型(図6Aのd)の生成が示唆される.
YC-1がヘム鉄の配位構造変化を引き起こすのはCO複合体に限ったことではなく,NO複合体でも見いだされている.YC-1が結合すると,対称性の高い5配位NO型のESRシグナル(図3C)は対称性の低いESRシグナル(図6B)に変換する7, 32).図3Cの対称性の高いESRシグナルを示す5配位NO型は低活性型であり,図6BのYC-1が結合した5配位NO型は高活性型であると考えられる7, 32).これは,ATPあるいはその誘導体が結合した場合にはESRシグナルの変化は見いだされず低活性型であるが,GTPあるいはその誘導体が結合するとYC-1結合型と類似の高活性型ESRシグナルに変換されることから裏づけられている7)(図6B).この変化は,Fe-NOの結合角の変化等の幾何学的な配位構造変化に由来するが,その相違は光吸収スペクトルでは区別できない.
それでは,YC-1はどのドメインに結合するのであろうか? 我々は,YC-1はヘテロ二量体あたり1分子結合すること,条件により若干異なるがその親和性は50 μM程度であることを見いだした27, 30).YC-1の誘導体であるBAY41-2272存在下では,先述したGMP-CPPの低親和性結合部位(偽触媒部位)への結合が完全に失われることから,我々は,YC-1ならびにBAY41-2272はC末端側の偽触媒部位に結合するモデルを提案した7).しかしながら,その後の欠損変異体を用いた実験結果は,その結合部位がβ鎖のN末端側に存在する可能性を強く示唆している33, 34).現在では我々の解釈は否定されているが,YC-1の結合部位とヌクレオチド結合部位の間に相互作用のあることは疑いない.実際,YC-1あるいはBAY41-2272により,基質であるGTPに対するKm値は50 μMから20 μMへと低下する.
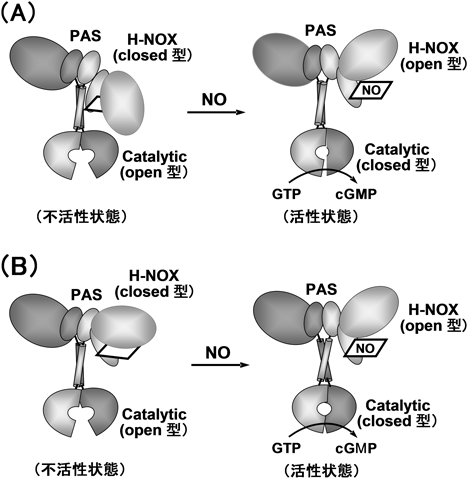
完全長のsGCの結晶構造はいまだに明らかにされてはいないが,single-particle electron microscopy(単粒子電子顕微鏡)により,おおよその立体構造が可視化されている35).さまざまな構造がとらえられているが,基本的には,sGCはハート形の小さなモデュール,それより大きなモデュール,それらをつなぐモデュールから構成されている(図7).小モデュールはαとβ鎖のC末端部位から構成される触媒部位であり,大モデュールは,α鎖のPASドメインとN末端ドメイン,β鎖のPASドメインとH-NOXドメインの両者のクラスターである.コイルドコイルヘリックスドメインが両モデュールをつないでいる.構造は柔軟性に富み,sGCはさまざまな構造をとりうる.それが,H-NOXドメインと触媒ドメインとの相互作用を可能にし,活性化の要因と解釈されている.
ドメイン間の相互作用は水素・重水素交換でも解析されている36).休止状態とNO結合型酵素の比較から,交換速度はヘムドメインのdistal side,触媒ドメインの接触部位ならびにコイルドコイル部分で著しい.これは,NO結合によりヘムドメインがopen型に,触媒ドメインはclosed型に移行するものと解釈されている.これらの結果に基づき,NOにより誘起されるシクラーゼ反応の機構として,ヘムドメインと触媒ドメイン間の接触が解除される大きなコンホメーション変化を伴う直接的な機構(図7A)と,PASとヘリックスドメイン間の接触の変化に起因するコイルドコイルのねじれを介したlong-rangeコンホメーション変化による機構(図7B)が提案されている.なお,触媒ドメインがopen型とclosed型の二つの状態をとることは,アデニル酸シクラーゼの結果から推定されたものであり,直接的な証拠は得られていない.完全長sGCの結晶構造に基づいたドメイン間の相互作用ならびにα鎖のN末端ドメインの機能について,今後の詳細な解析がまたれる.
本稿では,1990年代半ばから現在までの約20年にわたって明らかにされてきた可溶性グアニル酸シクラーゼの構造と機能の相関,特にNOによる活性化機構ならびにNO感知機構を中心に述べてきた.哺乳動物では,sGCがNOをシグナルとする唯一のセンサータンパク質と考えられるが,CO, O2あるいはH2Sをシグナル分子とするヘムを含むセンサータンパク質も数多く知られている37).しかしながら,他のセンサータンパク質に比べて,sGCのガス状分子に対する選択性の高さはきわだっており,その選択性を支える分子機構の詳細な解明がまたれる.本稿では紹介しなかったが,線虫の行動を支配するO2をシグナル分子とするヘムを含むグアニル酸シクラーゼも知られており,哺乳動物においても存在する可能性が高いと思われる.
紹介した我々の研究結果は,堀洋博士(元・阪大基礎工学部),城宜嗣理化学研究所主任研究員(現・兵庫県立大),朴三用理化学研究所研究員(現・横浜市大),尾林栄治理化学研究所研究員(現・島根医大),飯塚哲太郎博士(元・理化学研究所主任研究員)との共同研究によるものであり,深謝する.
引用文献References
1) Kamisaki, Y., Saheki, S., Nakane, M., Palmieri, J.A., Kuno, T., Chang, B.Y., Waldman, S.A., & Murad, F. (1986) J. Biol. Chem., 261, 7236–7241.
2) Yuen, P.S., Potter, L.R., & Garbers, D.L. (1990) Biochemistry, 29, 10872–10878.
3) Tesmer, J.J., Sunahara, R.K., Gilman, A.G., & Sprang, S.R. (1997) Science, 278, 1907–1916.
4) Allerston, C.K., von Delft, F., & Gileadi, O. (2013) PLoS ONE, 8, e57644.
5) Seeger, F., Quintyn, R., Tanimoto, A., Williams, G.J., Tainer, J.A., Wysocki, V.H., & Garcin, E.D. (2014) Biochemistry, 53, 2153–2165.
6) Sunahara, R.K., Beuve, A., Tesmer, J.G., Sprang, S.R., Garbers, D.L., & Gilman, A.G. (1998) J. Biol. Chem., 273, 16323–16328.
7) Yazawa, S., Tsuchiya, H., Hori, H., & Makino, R. (2006) J. Biol. Chem., 281, 21763–21770.
8) Surmeli, N.B., Muskens, F.M., & Marletta, M.A. (2015) J. Biol. Chem., 290, 15570–15580.
9) Beste, K.Y., Burhenne, H., Kaever, V., Stasch, J.-P., & Seifert, R. (2012) Biochemistry, 51, 194–204.
10) Ruiz-Stewart, I., Tiyyagura, S.R., Lin, J.E., Kazerounian, S., Pitari, G.M., Schulz, S., Martin, E., Murad, F., & Waldman, S.A. (2004) Proc. Natl. Acad. Sci. USA, 101, 37–42.
11) Suzuki, T., Suematsu, M., & Makino, R. (2001) FEBS Lett., 507, 49–53.
12) Wedel, B., Humbert, P., Harteneck, C., Foerster, J., Malkewitz, J., Bohme, E., Schultz, G., & Koesling, D. (1994) Proc. Natl. Acad. Sci. USA, 91, 2592–2596.
13) Zhao, Y., Schelvis, J.P., Babcock, G.T., & Marletta, M.A. (1998) Biochemistry, 37, 4502–4509.
14) Boon, E.M., Huang, S.H., & Marletta, M.A. (2005) Nat. Chem. Biol., 1, 53–59.
15) Pellicena, P., Karow, D.S., Boon, E.M., Marletta, M.A., & Kuriyan, J. (2004) Proc. Natl. Acad. Sci. USA, 101, 12854–12859.
16) Herzik, M.A. Jr., Jonnalagadda, R., Kuriyan, J., & Marletta, M.A. (2014) Proc. Natl. Acad. Sci. USA, 111, E4156–E4164.
17) Traylor, T.G. & Sharma, V.S. (1992) Biochemistry, 31, 2847–2849.
18) Stone, J.R. & Marletta, M.A. (1994) Biochemistry, 33, 5636–5640.
19) Makino, R., Matsuda, H., Obayashi, E., Shiro, Y., Iizuka, T., & Hori, H. (1999) J. Biol. Chem., 274, 7714–7723.
20) Deinum, G., Stone, J.R., Babcock, G.T., & Marletta, M.A. (1996) Biochemistry, 35, 1540–1547.
21) Zhao, Z., Brandish, P.E., Ballou, D.P., & Marletta, M.A. (1999) Proc. Natl. Acad. Sci. USA, 96, 14753–14758.
22) Lawson, D.M., Stevenson, C.E.M., Andrew, C.R., & Eady, R.R. (2000) EMBO J., 19, 5661–5671.
23) Martin, E., Berka, V., Sharina, I., & Tsai, A.-L. (2012) Biochemistry, 51, 2737–2746.
24) Yoo, B.-K., Lamarre, I., Martin, J.-L., Rappaport, F., & Negrerie, M. (2015) Proc. Natl. Acad. Sci. USA, 112, E1697–E1704.
25) Martin, E., Berka, V., Bogatenkova, E., Murad, F., & Tsai, A.-L. (2006) J. Biol. Chem., 281, 57836–27845.
26) Makino, R., Park, S.-Y., Obayashi, E., Iizuka, T., Hori, H., & Shiro, Y. (2011) J. Biol. Chem., 286, 15678–15687.
27) Makino, R., Yazawa, S., Hori, H., & Shiro, Y. (2012) Biochemistry, 51, 9277–9289.
28) Friebe, A., Schultz, G., & Koesling, D. (1996) EMBO J., 15, 6863–6868.
29) Denninger, J.W., Schelvis, J.P.M., Brandish, P.E., Zhao, Y., Babcock, G.T., & Marletta, M.A. (2000) Biochemistry, 39, 4191–4198.
30) Makino, R., Obayashi, E., Homma, N., Shiro, Y., & Hori, H. (2003) J. Biol. Chem., 278, 11130–11137.
31) Martin, E., Czarnecki, K., Jayaraman, V., Murad, F., & Kincaid, J. (2005) J. Am. Chem. Soc., 127, 4625–4631.
32) Derbyshire, E.R., Gunn, A., Ibrahim, M., Spiro, T.G., Britt, R.D., & Marletta, M.A. (2008) Biochemistry, 47, 3892–3899.
33) Purohit, R., Fritz, B.G., The, J., Issaian, A., Weichsel, A., David, C.L., Campbell, E., Hausrath, A.C., Rassouli-Taylor, L., Garcin, E.D., Gage, M.J., & Montfort, W.R. (2014) Biochemistry, 53, 101–114.
34) Yoo, B.K., Lamarre, I., Rappaport, F., Nioche, P., Raman, C.S., Martin, J.L., & Negrerie, M. (2012) ACS Chem. Biol., 7, 2046–2054.
35) Campbella, M.G., Underbakke, E.S., Pottera, C.S., Carraghera, B., & Marletta, M.A. (2014) Proc. Natl. Acad. Sci. USA, 111, 2960–2965.
36) Underbakke, E.S., Lavarone, A.T., & Marletta, A.M. (2013) Proc. Natl. Acad. Sci. USA, 110, 6777–6782.
37) Shimizu, T., Huang, D., Yan, F., Stranava, M., Bartosova, M., Fojtíkova, V., & Martínkova, M. (2015) Chem. Rev., 115, 6491–6533.
著者紹介Author Profile
 牧野 龍(まきの りゅう)
牧野 龍(まきの りゅう)立教大学名誉教授.立教大学客員研究員.理学博士(北海道大学).
略歴1947年北海道札幌市に生る.69年北海道大学理学部化学科卒業.74年同大学院理学研究科化学専攻博士課程修了(理学博士).同年米国イリノイ大学博士研究員.75年慶應義塾大学医学部助手.86年同専任講師.91年姫路工業大学理学部助教授.96年立教大学理学部化学科生命理学コース教授.2002年立教大学理学部生命理学科教授.12年立教大学名誉教授.
研究テーマと抱負ヘム酵素の構造と機能の解析.