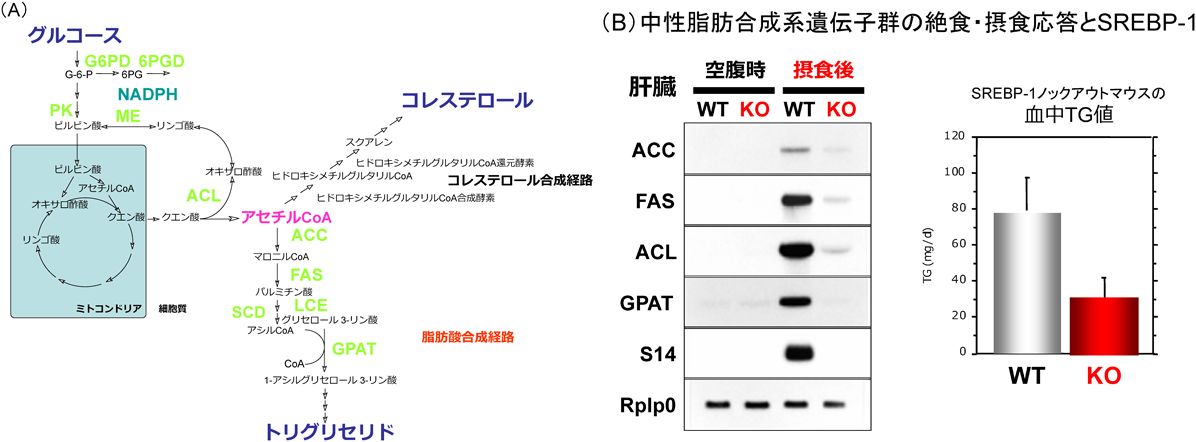
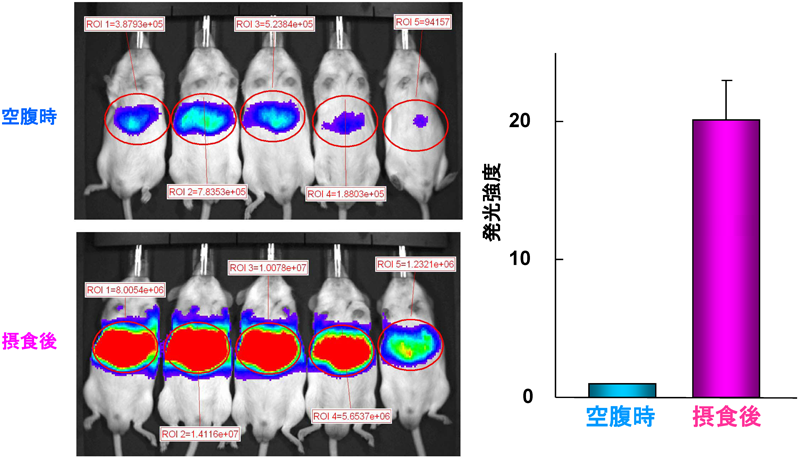
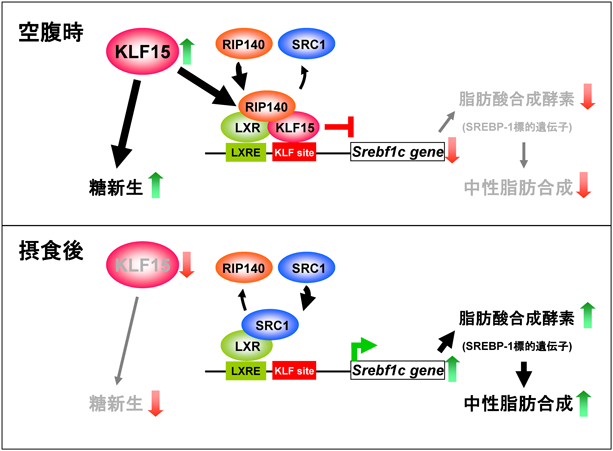
1) Takeuchi, Y., Yahagi, N., Aita, Y., Murayama, Y., Sawada, Y., Piao, X., Toya, N., Oya, Y., Shikama, A., Takarada, A., Masuda, Y., Nishi, M., Kubota, M., Izumida, Y., Yamamoto, T., Sekiya, M., Matsuzaka, T., Nakagawa, Y., Urayama, O., Kawakami, Y., Iizuka, Y., Gotoda, T., Itaka, K., Kataoka, K., Nagai, R., Kadowaki, T., Yamada, N., Lu, Y., Jain, M.K., & Shimano, H. (2016) Cell Reports, 16, 2373–2386.
2) Shimano, H., Yahagi, N., Amemiya-Kudo, M., Hasty, A.H., Osuga, J., Tamura, Y., Shionoiri, F., Iizuka, Y., Ohashi, K., Harada, K., Gotoda, T., Ishibashi, S., & Yamada, N. (1999) J. Biol. Chem., 274, 35832–35839.
3) Yahagi, N. & Shimano, H. (2005) in Understanding Lipid Metabolism with Microarrays and Other Omic Approaches (Berger, A. & Roberts, M.A. eds.), pp. 237–248, CRC Press.
4) Yokoyama, C., Wang, X., Briggs, M.R., Admon, A., Wu, J., Hua, X., Goldstein, J.L., & Brown, M.S. (1993) Cell, 75, 187–197.
6) Repa, J.J., Liang, G., Ou, J., Bashmakov, Y., Lobaccaro, J.M., Shimomura, I., Shan, B., Brown, M.S., Goldstein, J.L., & Mangelsdorf, D.J. (2000) Genes Dev., 14, 2819–2830.
7) Yoshikawa, T., Shimano, H., Amemiya-Kudo, M., Yahagi, N., Hasty, A.H., Matsuzaka, T., Okazaki, H., Tamura, Y., Iizuka, Y., Ohashi, K., Osuga, J., Harada, K., Gotoda, T., Kimura, S., Ishibashi, S., & Yamada, N. (2001) Mol. Cell. Biol., 21, 2991–3000.
8) Gray, S., Wang, B., Orihuela, Y., Hong, E.G., Fisch, S., Haldar, S., Cline, G.W., Kim, J.K., Peroni, O.D., Kahn, B.B., & Jain, M.K. (2007) Cell Metab., 5, 305–312.
9) Porstmann, T., Santos, C.R., Griffiths, B., Cully, M., Wu, M., Leevers, S., Griffiths, J.R., Chung, Y.L., & Schulze, A. (2008) Cell Metab., 8, 224–236.
10) Peterson, T.R., Sengupta, S.S., Harris, T.E., Carmack, A.E., Kang, S.A., Balderas, E., Guertin, D.A., Madden, K.L., Carpenter, A.E., Finck, B.N., & Sabatini, D.M. (2011) Cell, 146, 408–420.
11) Allmann, D.W. & Gibson, D.M. (1965) J. Lipid Res., 6, 51–62.
12) Yahagi, N., Shimano, H., Hasty, A.H., Amemiya-Kudo, M., Okazaki, H., Tamura, Y., Iizuka, Y., Shionoiri, F., Ohashi, K., Osuga, J., Harada, K., Gotoda, T., Nagai, R., Ishibashi, S., & Yamada, N. (1999) J. Biol. Chem., 274, 35840–35844.
13) Takeuchi, Y., Yahagi, N., Izumida, Y., Nishi, M., Kubota, M., Teraoka, Y., Yamamoto, T., Matsuzaka, T., Nakagawa, Y., Sekiya, M., Iizuka, Y., Ohashi, K., Osuga, J., Gotoda, T., Ishibashi, S., Itaka, K., Kataoka, K., Nagai, R., Yamada, N., Kadowaki, T., & Shimano, H. (2010) J. Biol. Chem., 285, 11681–11691.
14) Sekiya, M., Yahagi, N., Matsuzaka, T., Takeuchi, Y., Nakagawa, Y., Takahashi, H., Okazaki, H., Iizuka, Y., Ohashi, K., Gotoda, T., Ishibashi, S., Nagai, R., Yamazaki, T., Kadowaki, T., Yamada, N., Osuga, J., & Shimano, H. (2007) J. Lipid Res., 48, 1581–1591.
15) Nishi-Tatsumi, M., Yahagi, N., Takeuchi, Y., Toya, N., Takarada, A., Murayama, Y., Aita, Y., Sawada, Y., Piao, X., Oya, Y., Shikama, A., Masuda, Y., Kubota, M., Izumida, Y., Matsuzaka, T., Nakagawa, Y., Sekiya, M., Iizuka, Y., Kawakami, Y., Kadowaki, T., Yamada, N., & Shimano, H. (2017) FEBS Lett., 591, 965–978.
16) Yahagi, N., Shimano, H., Hasty, A.H., Matsuzaka, T., Ide, T., Yoshikawa, T., Amemiya-Kudo, M., Tomita, S., Okazaki, H., Tamura, Y., Iizuka, Y., Ohashi, K., Osuga, J., Harada, K., Gotoda, T., Nagai, R., Ishibashi, S., & Yamada, N. (2002) J. Biol. Chem., 277, 19353–19357.