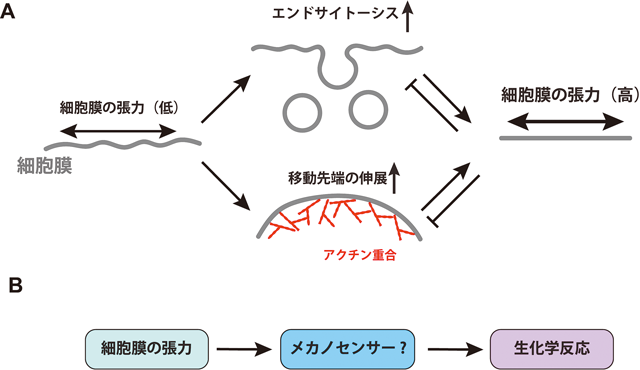
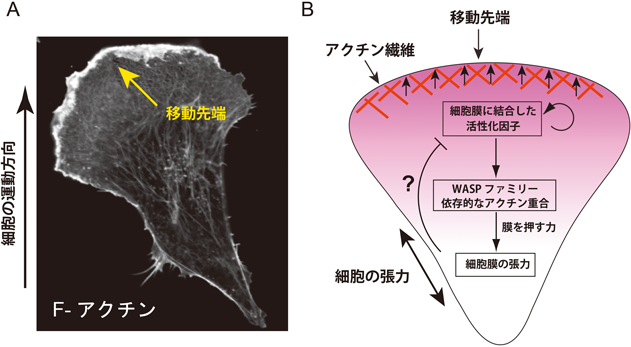
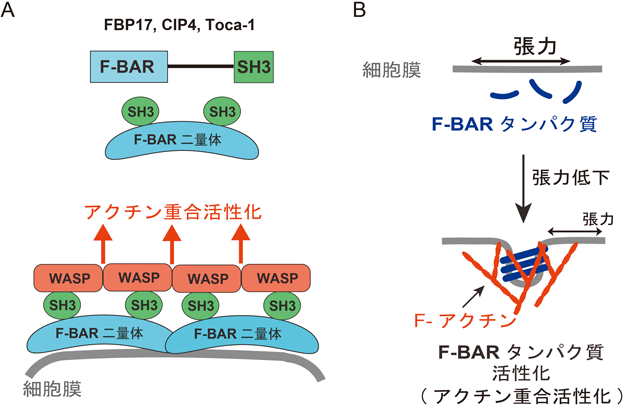
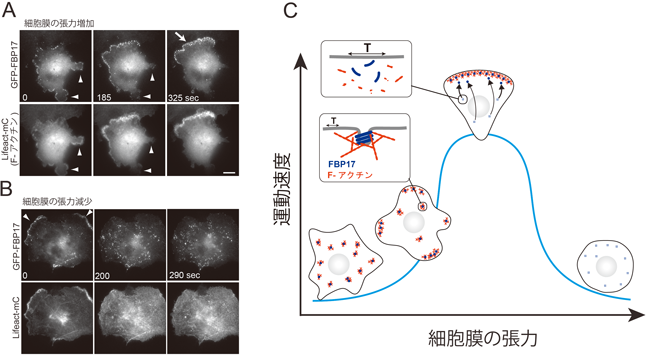
5) Tsujita, K., Suetsugu, S., Sasaki, N., Furutani, M., Oikawa, T., & Takenawa, T. (2006) J. Cell Biol., 172, 269–279.
6) Shimada, A., Niwa, H., Tsujita, K., Suetsugu, S., Nitta, K., Hanawa-Suetsugu, K., Akasaka, R., Nishino, Y., Toyama, M., Chen, L., Liu, Z.J., Wang, B.C., Yamamoto, M., Terada, T., Miyazawa, A., Tanaka, A., Sugano, S., Shirouzu, M., Nagayama, K., Takenawa, T., & Yokoyama, S. (2007) Cell, 129, 761–772.
9) Rohatgi, R., Ma, L., Miki, H., Lopez, M., Kirchhausen, T., Takenawa, T., & Kirschner, M.W. (1999) Cell, 97, 221–231.
12) Neilson, M.P., Veltman, D.M., van Haastert, P.J., Webb, S.D., Mackenzie, J.A., & Insall, R.H. (2011) PLoS Biol., 9, e1000618.
14) Weiner, O.D., Marganski, W.A., Wu, L.F., Altschuler, S.J., & Kirschner, M.W. (2007) PLoS Biol., 5, e221.
15) Veltman, D.M., King, J.S., Machesky, L.M., & Insall, R.H. (2012) J. Cell Biol., 198, 501–508.
19) Gauthier, N.C., Fardin, M.A., Roca-Cusachs, P., & Sheetz, M.P. (2011) Proc. Natl. Acad. Sci. USA, 108, 14467–14472.
20) Lieber, A.D., Yehudai-Resheff, S., Barnhart, E.L., Theriot, J.A., & Keren, K. (2013) Curr. Biol., 23, 1409–1417.
21) Houk, A.R., Jilkine, A., Mejean, C.O., Boltyanskiy, R., Dufresne, E.R., Angenent, S.B., Altschuler, S.J., Wu, L.F., & Weiner, O.D. (2012) Cell, 148, 175–188.
23) Chang, G., Spencer, R.H., Lee, A.T., Barclay, M.T., & Rees, D.C. (1998) Science, 282, 2220–2226.
29) Takei, K., Slepnev, V.I., Haucke, V., & De Camilli, P. (1999) Nat. Cell Biol., 1, 33–39.
30) Razzaq, A., Robinson, I.M., McMahon, H.T., Skepper, J.N., Su, Y., Zelhof, A.C., Jackson, A.P., Gay, N.J., & O’Kane, C.J. (2001) Genes Dev., 15, 2967–2979.
31) Lee, E., Marcucci, M., Daniell, L., Pypaert, M., Weisz, O.A., Ochoa, G.C., Farsad, K., Wenk, M.R., & De Camilli, P. (2002) Science, 297, 1193–1196.
32) Peter, B.J., Kent, H.M., Mills, I.G., Vallis, Y., Butler, P.J., Evans, P.R., & McMahon, H.T. (2004) Science, 303, 495–499.
34) Kamioka, Y., Fukuhara, S., Sawa, H., Nagashima, K., Masuda, M., Matsuda, M., & Mochizuki, N. (2004) J. Biol. Chem., 279, 40091–40099.
35) Itoh, T., Erdmann, K.S., Roux, A., Habermann, B., Werner, H., & De Camilli, P. (2005) Dev. Cell, 9, 791–804.
36) Frost, A., Perera, R., Roux, A., Spasov, K., Destaing, O., Egelman, E.H., De Camilli, P., & Unger, V.M. (2008) Cell, 132, 807–817.
37) Henne, W.M., Boucrot, E., Meinecke, M., Evergren, E., Vallis, Y., Mittal, R., & McMahon, H.T. (2010) Science, 328, 1281–1284.
38) Senju, Y., Itoh, Y., Takano, K., Hamada, S., & Suetsugu, S. (2011) J. Cell Sci., 124, 2032–2040.
41) Ho, H.Y., Rohatgi, R., Lebensohn, A.M., Le, M., Li, J., Gygi, S.P., & Kirschner, M.W. (2004) Cell, 118, 203–216.
43) Tsujita, K., Kondo, A., Kurisu, S., Hasegawa, J., Itoh, T., & Takenawa, T. (2013) J. Cell Sci., 126, 2267–2278.
44) Padrick, S.B., Cheng, H.C., Ismail, A.M., Panchal, S.C., Doolittle, L.K., Kim, S., Skehan, B.M., Umetani, J., Brautigam, C.A., Leong, J.M., & Rosen, M.K. (2008) Mol. Cell, 32, 426–438.
45) Dai, J., Sheetz, M.P., Wan, X., & Morris, C.E. (1998) J. Neurosci., 18, 6681–6692.
47) Chander, H., Truesdell, P., Meens, J., & Craig, A.W. (2012) Oncogene, 32, 3080–3090.
48) Giannone, G., Dubin-Thaler, B.J., Dobereiner, H.G., Kieffer, N., Bresnick, A.R., & Sheetz, M.P. (2004) Cell, 116, 431–443.
49) Giannone, G., Dubin-Thaler, B.J., Rossier, O., Cai, Y., Chaga, O., Jiang, G., Beaver, W., Dobereiner, H.G., Freund, Y., Borisy, G., & Sheetz, M.P. (2007) Cell, 128, 561–575.