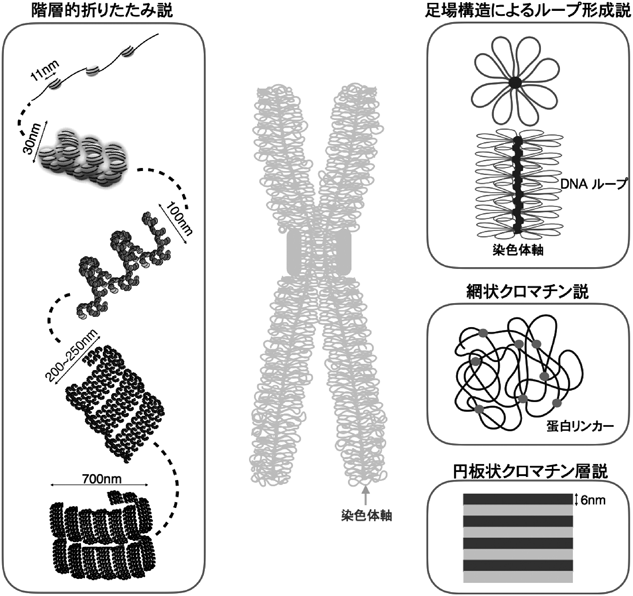
ドイツの細胞学者Walther Flemmingが,細胞核内にアニリンで強く染まる構造を見いだし,クロマチンと名づけたのが1879年のことである1).Flemmingはクロマチンが細胞分裂の際に棒状の構造物となり,それが二つに分かれて娘細胞に分配されていく過程の精密で美しいスケッチを残した2).その後多くの学者がこの棒状の構造物,すなわち染色体の謎解きに取り組むことになる.1953年にWatsonとClickはDNAの二重らせん構造を3),1974年にKornbergらはヒストンタンパク質にDNAが巻きついたヌクレオソーム構造を発見した4).1970年代以降は,X線回折パターン,電子顕微鏡像などから,より高次のクロマチン線維の折りたたまり,すなわち染色体凝縮の仮説が導かれるようになっていったが,今日に至ってもなお,新たなモデルが提唱されるように,染色体凝縮の謎解きが続いている.染色体の形状や,核型分析の際の染色体上のバンドパターンは,染色体ごとに決まっていることから,染色体は一定の原則や規則性に沿って凝縮すると考えてまず間違いない.以下に,これまでに提唱された主だった染色体凝縮のモデルを示す(図1).おそらく実際には,正解は一つではなく,これらの要素が複合的に用いられている可能性がある.
1)階層的折りたたみ説(hierarchical folding model)
11 nm幅のヌクレオソームに巻いたDNAが,リンカーヒストンであるヒストンH1を伴い規則正しくらせん状に折りたたまれて30 nm幅のクロマチンファイバーとなり,その30 nmファイバーがさらに高次のらせん状に折りたたまれながら段階的に径が大きくなって,最終的に姉妹染色分体を作るというモデルである5).教科書でも説明される広く受け入れられているモデルである.一方で近年,急速凍結したM期細胞の電子顕微鏡観察によれば,直径30 nm程度の線維状構造は検出されず,30 nmファイバーの存在の根拠となったX線構造解析のデータは,染色体周囲に存在するリボソームRNAを検出したことに由来するのではないかという議論がある6).また,DNA染色方法を改良した最新の電子顕微鏡像の解析でも,間期と分裂期でクロマチン繊維の秩序だった構造変化は認めず,クロマチン繊維の凝縮密度の差によって間期クロマチンと分裂期染色体の形態の違いが生じることが観察されており7),細胞内では階層的なクロマチン構築は存在しないことが示唆されている.
2)足場構造によるループ形成説(scaffold/radial-loop model)
M期染色体を脱ヒストン化処理し,電子顕微鏡で観察すると,染色体の中心に非ヒストン性のタンパク質による軸状の構造が検出された8).この構造をスキャフォールド(足場)と呼び,文字どおりそこを足場にしてDNAのループ構造が放射状に広がるようすが観察されたことから導かれたモデルである.スキャフォールドには後に詳述するコンデンシンやトポ2,KIF4Aといった染色体構築に重要な役割を果たすタンパク質が局在している.
3)網状クロマチン説(chromatin network model)
染色体は,長軸方向に引き延ばす力を加えると,もとの形状に戻ろうとする.この弾力性はヌクレアーゼ処理によって失われたが,プロテアーゼで穏やかに処理した場合は,引き延ばしを維持するための力は徐々に減少したものの弾力性そのものは失われなかった9).これらの結果から,M期染色体を形作るのはスキャフォールド・タンパク質などの足場構造に基づくものではなく,クロマチン自体の機械的な連続が主成分ではないかとする,クロマチンネットワークモデルが提唱された.
4)円板状クロマチン層説(thin layers stack model)
核型解析で用いられるG-band法などの染色体バンドや,姉妹染色分体交換や転座箇所にみられる染色体内部の境界面は,染色体長軸に対して垂直に形成され,また境界面が明瞭に認識できる10).これらの観察は,上記三つのモデルでは明快な説明が難しく,染色体は約6 nmの薄さの円盤状のクロマチン層が積み重なって形成されているという説である.
1)コンデンシンの発見
上記のようにLaemmliらが,脱ヒストン化した染色体を電子顕微鏡で観察して,足場構造によるループ形成説を報告してから8),以後の染色体研究ではスキャフォールド・タンパク質の同定が重要な課題の一つとなっていった.その中には,後にSMC(structural maintenance of chromosomes)と総称されるATPase活性を持つタンパク質やKleisinタンパク質,あるいはトポイソメラーゼIIの発見があり,これらの発見に日本人研究者が大きく貢献した.たとえば,仁木らは1991年に大腸菌でMukB(SMCに相当)を11),斉藤らは1994年にニワトリのScII(SMC2)を12)それぞれクローニングした.また,平野らは1994年にアフリカツメガエルのXCAP-C/E(SMC2/SMC4)のヘテロ二量体を同定した13).決定的な発見は,1997年の平野らによるコンデンシン複合体の同定である14).平野らはカエル卵抽出液で再構成した染色体から,XCAP-C/Eが三つのタンパク質XCAP-D2/G/Hと五量体を形成し,このコンデンシンと名づけられた複合体が染色体凝縮活性を持つことを突き止めた.コンデンシンの発見により染色体の分子細胞生物学的研究が本格的に幕を開けた.
2)コンデンシンの構造
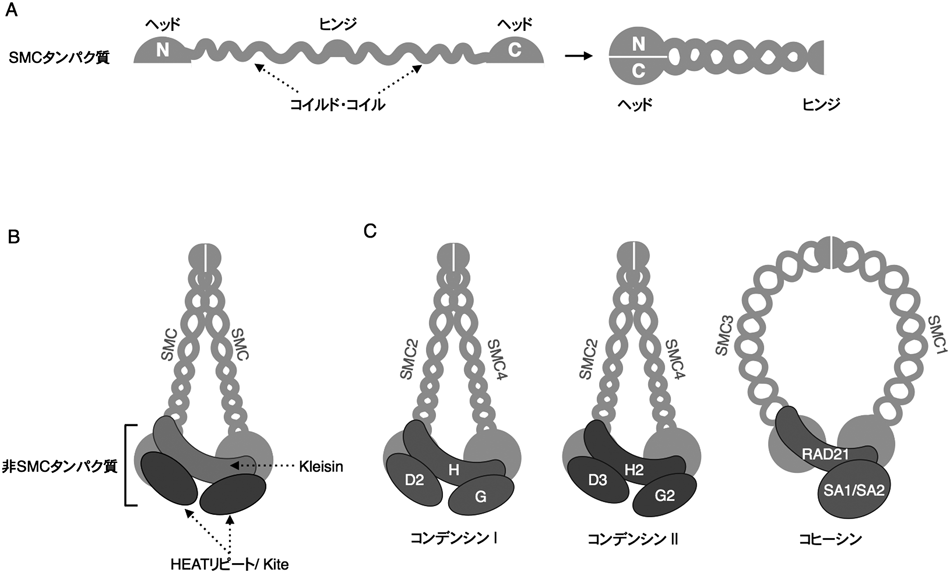
コンデンシン複合体あるいはそのプロトタイプは三つの生物ドメイン,すなわち真正細菌,古細菌,真核生物のすべてに存在し,生物進化の初期の段階から,複製されたDNAを分配する役割を担ってきたと考えられている15).コンデンシン複合体は二つのSMCタンパク質と三つの非SMCタンパク質で構成されており,後者はKleisinサブユニットと,二つのHEATリピートタンパク質(真核生物)またはKiteタンパク質(原核生物)からなる(図2).Kiteは近年新たに提唱された名称で,Kleisin interacting winged-helix(WH)tandem elementsの略であり,DNA結合モジュールとして知られるWHドメインを有するのが特徴である16).
SMCタンパク質は中央にヒンジ領域を有し(図2A),そこでタンパク質が二つに折れ曲がり,アミノ末端(N末端)とカルボキシル末端(C末端)が一つとなって,ATPase活性を持つヘッド・ドメインを形成する.ヒンジ領域とヘッド・ドメインの間は約50 nmの長いコイルド・コイル領域からなり,二つのSMCタンパク質は互いのヒンジ領域で結合しV字型の二量体を形成する.SMCタンパク質は原核生物ではホモ二量体であるが(枯草菌はSMC/SMC,大腸菌はMukB/MukB),真核生物ではSMC2/SMC4のへテロ二量体を形成している(図2B).
KleisinサブユニットのN末端は,一方のSMCヘッド・ドメイン近傍に位置するコイルド・コイルのneck領域に,C末端は他方のSMCヘッド・ドメインの先端にあるcap領域にそれぞれ結合する17).その結果,SMC二量体とKleisinサブユニットの三つのタンパク質でリングを形成する(リングといっても形状は桿状に近い).このリング構造とATPaseの活性により,後述するようにDNAと相互作用する.二つのHEATタンパク質あるいはKiteタンパク質はKleisinサブユニットに結合し,コンデンシンに機能を付与する重要な役割を担う.多くの真核生物には,非SMCタンパク質の異なる2種類のコンデンシン複合体(コンデンシンIとコンデンシンII)が存在することが知られている(図2C)18, 19).これらは両者に共通のSMC2/4と,それぞれに固有の三つの非SCMタンパク質から構成されており,後述するように細胞内局在や機能に相違があることがわかっている.
3)コンデンシンの局在
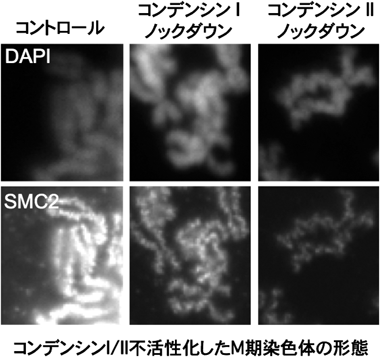
ヒトには2種類のコンデンシンが存在し,それぞれ細胞周期で異なった動態を示す19, 20).コンデンシンIは間期の間は細胞質に局在し,前中期に核膜が崩壊して初めて染色体に結合して,染色体構造の中心を縦走するタンパク質の足場構造である“軸”と呼ばれる部位に濃縮する(図1).一方,コンデンシンIIは,間期から核内に局在し,S期のDNA複製に伴いクロマチンに結合し21),分裂前期になると染色体の形成とともに染色体の“軸”に濃縮する.このような局在の違いから,コンデンシンIとIIがそれぞれ異なる役割を担っていることが推測される(図3).つまり,コンデンシンIIによる分裂前期での染色体凝縮は,核膜崩壊後にコンデンシンIが加わることで,さらなる高次構造の形成が起こり,染色体の構築が完成すると考えられる.
近年,ChIP-seq解析(クロマチン免疫沈降とDNA配列解読を組み合わせた解析)によって,ゲノム上のコンデンシン結合部位をマッピングできるようになった.コンデンシンは広く染色体上に分布するが,とりわけ,セントロメアDNA, RNAポリメラーゼI(pol I)に転写されるrRNA遺伝子の繰り返し配列,pol IIIに転写されるtRNA遺伝子,5S rRNA遺伝子,pol IIに転写される遺伝子に,より多く結合していることが示されている22–27).また,興味深いことに,コンデンシンは転写活性化領域に生じる一本鎖DNAに優先的に結合することが報告されており25, 28),これは後述するDNAとの結合様式を説明する上でつじつまが合う知見である.また,コンデンシンはM期に発現が誘導される標的遺伝子の転写調節領域間の相互作用にも関与していることが酵母におけるChIA-PET解析(タグの配列解析を用いたクロマチン相互作用の解析法)により示されている26).
4)コンデンシンのDNA結合様式
コンデンシン複合体がどのようにDNAと結合しているのかは,コンデンシン研究の重要課題の一つである.SMC複合体の一つ,コヒーシン(図2B)は,そのリングの内部にDNAを通すようにしてクロマチンと結合する(このような結合様式をトポロジカルな結合と表現する).コンデンシンにおいても枯草菌や出芽酵母を用いた研究により,コンデンシンリングの内部にDNAを通す形でトポロジカルに結合することが示唆された29, 30).実際,出芽酵母のコンデンシンと環状DNAの結合はそのDNAを線状化することや,コンデンシンのリング構造の一部にタンパク質分解酵素によって切れ目を導入することで結合が外れることが示されている.
構造学的な解析によると,SMC2/4のヒンジ領域の構造は種を超えて保存性が高く,同部にDNAが結合することがわかっている31, 32).さらには,このヒンジ領域とDNAの結合は,一本鎖DNAの方が二本鎖DNAよりも強いことが示された33).SMCのヒンジ領域どうしは接合部を作る.コヒーシンの場合,この接合部がDNAのリング内への入り口になると考えられているが,コンデンシンの場合は,ヒンジ領域近傍のコイルド・コイルどうしが結合した状態が維持されるため,DNAの入り口にはなりにくいようだ34).
ヒンジ領域以外では,HEATリピートタンパク質に二本鎖DNAが結合することが示されている35).また,非SMCサブユニットを伴う五量体ではDNA存在下においてヘッド・ドメインのATPase活性が,SMC2/4の二量体に比べて10倍程度高まる.つまり,五量体では,HEATタンパク質にDNAが結合することで,ATPase活性が上昇することが示唆される.またHEATタンパク質を欠いたコンデンシンはM期に染色体上への局在が阻害される.したがって,HEATタンパク質へのDNA結合とSMCのATPase活性とコンデンシンの染色体局在は相互に関連していると考えられる.
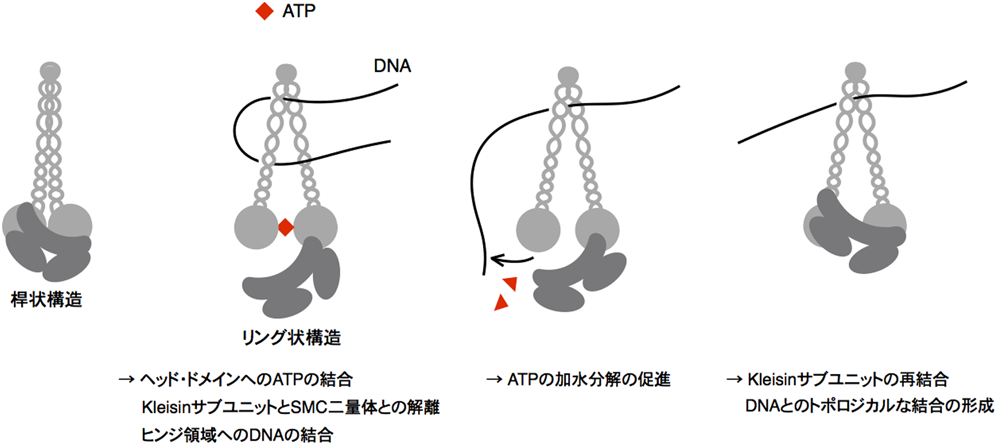
さらにSMC二量体内に存在するATPase活性の役割については,枯草菌の研究で,SMCのヘッド・ドメインへのATPの結合がヒンジ領域へのDNAの結合を促進することや,ヒンジ領域へのDNAの結合により,ヘッド・ドメインのATP加水分解が促進されることが示されている33, 36).これらの観察から,ヒンジ領域とヘッド・ドメインはそれぞれDNAやATPと結合すると,互いにコイルド・コイルを伝って立体構造が変化するような仕組みがあることが想定される(図4).
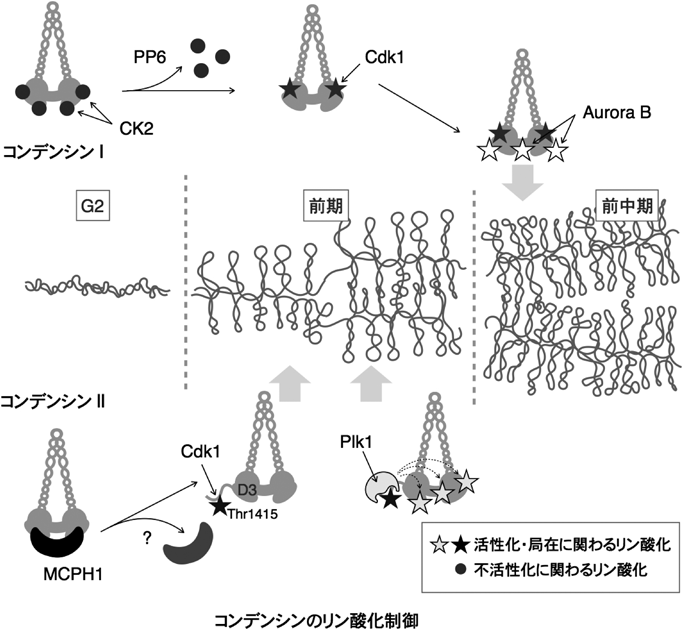
5)コンデンシンのリン酸化制御
コンデンシンの機能は,複数のM期キナーゼ,およびホスファターゼによって制御されている(図5).リン酸化がどのようにしてコンデンシンの活性や局在を調節するのか,その機構の詳細は未解明だが,コンデンシンのリン酸化は,M期の染色体凝縮を誘導するために必須の修飾であることが知られている.
a.M期サイクリン依存性キナーゼ(Cdk1)
酵母や後生動物のコンデンシンはM期にCdk1によりリン酸化される.in vitroにおいてコンデンシンのCAP-D2とCAP-HサブユニットがCdk1によるリン酸化を受けると,コンデンシンの二本鎖DNAにねじれを導入する活性(super-coiling活性)がきわだって上昇する37).Cdk1によりsuper-coiling活性を高めることが,M期で染色体の凝縮を誘導する背景として重要であると考えられる.我々は,分裂前期にCdk1がコンデンシンIIのCAP-D3のC末端部のThr1415位をリン酸化することが染色体凝縮の引き金になることを見いだした38).このThr残基は,核膜が壊れて染色体を分配する生物種で保存されており,活性の上がったCdk1が染色体凝縮を起こすために重要なリン酸化修飾であると予測される.
一方,酵母では,非SMCではなく,SMC4がCdk1によりリン酸化されることが知られている.出芽酵母SMC4のN末端にはリン酸化部位が複数存在するので,Cdk1の活性が低いM期の初期からリン酸化を受けやすく,それゆえにコンデンシンを活性化できるのではないかと予測されている39).また分裂酵母では,SMC4のリン酸化が,コンデンシンの核内移行を促すと報告されている40).哺乳類細胞にこれらに相当する機構は知られていないが,コンデンシンがCdk1の直接の基質となってM期の染色体凝縮を誘導するという関係は共通している.
b.Poloキナーゼ
コンデンシンの活性化には,Cdk1に加えて,Poloキナーゼが重要な役割を担っていることが出芽酵母の研究からわかってきた.in vitroでCdk1によって上昇するコンデンシンのsuper-coiling活性は,Poloが加わることによってさらに高い活性が検出されたことによる41).興味深いことにヒト細胞では,Cdk1とPoloキナーゼの一つPlk1(Polo-like kinase 1)が直接関連していることがわかっている.すなわち,上述したCdk1がリン酸化したCAP-D3のThr1415位を,Plk1のリン酸化ペプチド結合ドメインであるPolo-box domainが認識して,Plk1がCAP-D3に結合する結果,Plk1がコンデンシンII複合体に複数のリン酸化を誘導し,コンデンシンの働きを強める38).つまり,M期にはCdk1とPlk1がコンデンシンIIをリン酸化して,間期レベルからは少なくとも二段階活性が上がり,前期で染色体凝縮を進めると考えられる.
c.Aurora Bキナーゼ
Aurora BはPolo同様に高度に保存されたM期キナーゼであり,M期染色体構成因子をリン酸化することにより,クロマチンへの結合および解離を調節する.コンデンシンIはその制御下にあり,Aurora Bの活性を阻害した細胞では,核膜崩壊後の染色体局在が著明に減少する42).M期の細胞のAuroraB活性を阻害すると速やかにコンデンシンが染色体より消失することから,ターンオーバーの速いコンデンシンIがクロマチンに結合するためには常にAurora Bの活性を必要としていることがわかる.カエル卵抽出液を用いた検討では,Aurora BによるコンデンシンIの染色体局在はCdk1活性に依存しないことが示されており43),Aurora Bにより特異的に受けるリン酸化によってコンデンシンIのクロマチンとの親和性を変えるのではないかと考えられる.リン酸化されたコンデンシンIのサブユニットはSDS-PAGEで泳動度の遅いバンドとして捉えることができるが,これを用いるとAurora BはCAP-D2, CAP-G, CAP-Hのリン酸化に関与していることが示唆される42).
出芽酵母においてもAurora B(Ipl1)とコンデンシンIとの関わりは指摘されている.出芽酵母においては,中期までよりも後期での凝縮が目立つが,この過程にはAurora BによるコンデンシンIのリン酸化が必要であると報告されている44).後述するとおりヒト細胞においても,染色体の凝縮度は後期で最も高くなるが,この後期の凝縮はAurora B活性を必要とする45).後期という染色体を分配する時期においてAurora BとコンデンシンIの関係性は酵母からヒトまで保存されている点が興味深い.
d.ホスファターゼ
ヒトのコンデンシンIは間期にcasein kinase 2(CK2)によってリン酸化を受けていることが知られているが,このリン酸化はM期キナーゼによるものとは異なり,コンデンシンの活性を逆に抑制する46).したがってM期では,CK2によりリン酸化されたコンデンシンIは脱リン酸化される必要があり,PP6ホスファターゼによって特異的にリン酸基が外される47).PP6とコンデンシンIのノックダウン実験では,ともに類似した染色体構築異常を示すことからも,PP6はCK2と特異的に拮抗してコンデンシンIの働きを制御していることがわかる.
6)コンデンシンと相互作用する酵素
a.トポイソメラーゼ2α
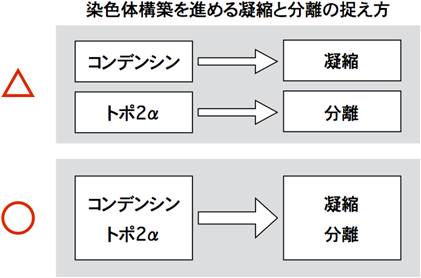
染色体の屋台骨となる軸構造にはコンデンシンに加えてトポイソメラーゼ2α(トポ2)が濃縮している48).このコンデンシンとトポ2の軸への局在は相互に依存するなど機能的には強く関連していると考えられている49).酵母においてプラスミドDNAを用いた実験で,コンデンシンによるsuper-coiling活性が,DNA間の絡まりを解除するトポ2の働きを促進することが示されている50, 51).ヒト細胞においても,染色体の凝縮と姉妹染色分体の分離は表裏一体で同時に進むので,その進行にはコンデンシンとトポイソメラーゼの両方が必要であることがわかっている52).
そもそもトポ2は,一つのDNA鎖に一時的な二本鎖切断を入れ,そこにもう一つのDNA鎖を通すという活性を持つ酵素である.そのため,トポ2は姉妹染色分体DNA間の絡まりを解く方向にも,作る方向にも働きうる.しかし,実際にM期で絡まりを解くこと(デカテネーション)を促進して姉妹染色分体の解離を進めるのは,トポイソメラーゼとともにコンデンシンが働くためとの見方ができる53).つまり,コンデンシンは染色体凝縮を,トポ2は姉妹染色分体の解離をそれぞれ促進し,両者が協調して染色体構築を進めるという従来の捉え方よりも,凝縮と解離という不可分のプロセスが,コンデンシンとトポ2によって進められると理解するのが妥当と思われる(図6).
実際にコンデンシンとトポイソメラーゼが,機能的な単位を作っているという仮説は,これまでにもさまざまな実験系によって支持されている.ショウジョウバエのBarren(CAP-H)とTop254),大腸菌のMukBとTopo IV(Top2に相当)55, 56)は,ともに相互作用してデカテネーションを促進することが示されている.したがって,コンデンシンとトポ2の働きは,染色体凝縮の根幹にあると考えられ,両者の相互作用の明快な説明が待たれる.
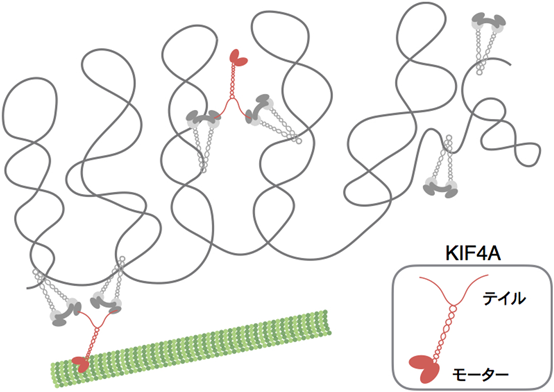
b.クロモキネシンKIF4A
KIF4Aは多くの真核生物が持つキネシンで,積荷を載せて微小管上をプラス端方向に移動するモーター分子である.KIF4Aはコンデンシンと同様にM期染色体の軸構造に局在し,染色体の構築に関与する57).KIF4AのC末端にあるテイル・ドメインは積荷となるいろいろなタンパク質と結合するが,我々はこのテイル・ドメインがコンデンシンIのCAP-GサブユニットとM期特異的に結合することを見いだした58).CAP-Gとの結合ができないテイル・ドメインに点変異を持つ細胞では,コンデンシンIの染色体軸への局在が損なわれ,コンデンシンIによる染色体構築が進まず,セントロメア構造が脆弱になった.さらに,KIF4Aのモーター活性を阻害した変異体を発現する細胞では,コンデンシンIとの結合は保たれていたにも関わらず,コンデンシンIの染色体軸への濃縮が起こらなかった.すなわち,コンデンシンIが軸構造に濃縮し,染色体を構築するためには,KIF4Aと結合し,そのモーター活性を使っていることが示唆され,キネシンのモーター活性が関与する染色体構築制御という新たな局面が見いだされた.
KIF4Aは細胞内で二量体を形成することから,理論上はKIF4AにはコンデンシンIとの結合部位が2か所あることになる.そこで,DNAと結合したコンデンシンIを2分子たぐり寄せるようにKIF4Aが結合することで,DNAにループ構造が形成されるのではないかと筆者らは考えている.KIF4Aのモーター活性によりコンデンシンIは染色体軸へ濃縮するとともに,ループ構造が完成するとの仮説が導き出される(図7).
1)染色体凝縮の定量的な顕微鏡解析
M期の進行に伴う染色体の凝縮程度を明らかにすべく,蛍光標識ヒストン発現細胞のライブイメージング解析がなされている.一つの細胞で染色体の体積変化を追うと,期待どおり前期より体積が減少し始めたが,後期において最も強く凝縮することが示された45).この後期の凝縮は,この時期に染色体の局在量が最大となるコンデンシンIが関与するに違いない.分離した姉妹染色分体が両極へ動きつつ染色体の腕部を収縮させることで娘細胞への分配を確実にしていると考えられる65).興味深いことに,この後期の凝縮には,微小管の動態やAurora B活性にも依存していることが示されている.
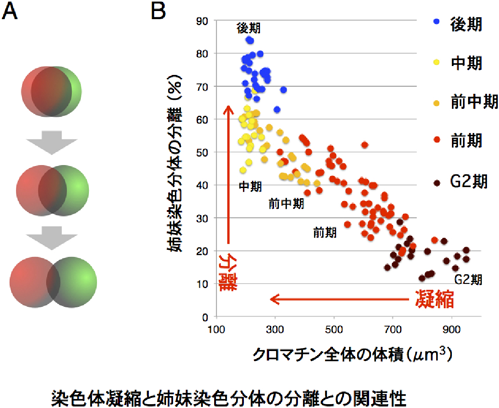
凝縮の進んだヒトの中期染色体では姉妹染色分体を別々に識別できる.凝縮と姉妹染色分体の分離過程との関連性を調べるために,2種類のヌクレオシド誘導体によって姉妹染色分体を別々の蛍光色に標識した細胞を用いて,姉妹染色分体が重なり合う領域を抽出することによって,分離過程の定量的解析がなされた52).この実験系を用いることで,前期において大部分の姉妹染色分体の分離が完了していることがわかった(図8).前期での分離はトポ2の機能阻害によって抑えられたが,コンデンシンIIの機能阻害によっても抑制された.つまり,前期における染色体の凝縮は,姉妹染色分体の分離を伴って進行しており,この過程はコンデンシンIIとトポ2によって協調的に制御されていることが示唆された.前期での変化を詳細に観察すると,凝縮が一度弛緩して膨張するようにみえるポイントの存在が指摘されており,姉妹染色分体を結合していた接着の解除による染色体の形態変化であろうと予測されている66).
また分裂酵母においては,染色体上の蛍光標識した2点間の距離によって凝縮度を予測するという実験により,コンデンシン,トポ2に加えて,Auroraキナーゼの必要性が示され,これらの分子が生物種を超えて重要な役割を担っていると考えられている67).さらに,ヒト細胞の染色体の体積測定を自動化することにより,M期の,特に前期での染色体凝縮に関わる分子が網羅的に調べられた.その結果,コンデンシンII以外でこれまで凝縮との関わりが知られていなかった分子が同定されており,今後の解析が待たれる68).
2)染色体軸構造の超微構造
従来の光学解像限界を超えた顕微鏡を活用して,染色体のより詳細な構造の観察が進められている.ヒト細胞を低張液処理することによって分離した染色体を,超解像度顕微鏡(3D-SIM法)と集束イオン/電子ビーム加工観察装置(FIB/SEM法)で解析したところ,染色体軸ではコンデンシンやトポ2といった軸構成分子が,DNAの二本鎖のように,二本の軸がよじれた構造をとっていることが示されている69).DNAのループ構造をいかに束ねて高次構造を構築するのかを解く上で示唆に富む知見である.
3)新しいゲノム学的解析による知見
近年,細胞内でDNAの領域と領域がどのような空間的距離をとるのかを網羅的に調べる3C(chromosome conformation capture)法,およびHi-Cと総称されるその変法が開発された.これを用いた解析によって,間期のクロマチンには数メガ塩基長の区画がありその中にTAD(topologically associated domain:TAD)と呼ばれる機能的まとまり(ドメイン)が作られることが明らかになりつつある70).ところがM期になると,こうした区画やドメインはすべて消失して,染色体のどの領域間の距離もほぼ一様になることが示された71).このデータに基づくクロマチン線維のシミュレーション解析は,M期の染色体は~80 kb程度のループを連続的に形成しかつそのループを束ねている足場が線状に並んだ構造をとっていると予測している.これらの結果は,クロマチン線維にはいくつかの階層があるという説を覆し,染色体は一様にその中心に足場を持つループ構造をとっていることを示唆するものである.染色体はコンデンシンおよびトポ2が濃縮する軸構造によって形作られている,という捉え方をさらに裏づける知見である.
1)小頭症
コンデンシンとヒトの疾患との関連は,小頭症において見いだされた.MCPH1は常染色体劣性遺伝である原発性小頭症の原因遺伝子の一つであるが,これに変異を持つ細胞のM期での染色体凝縮は通常よりも早いタイミングで起こることが報告された72, 73).その後,MCPH1はコンデンシンIIと特異的に結合してその働きを抑えていることが示された.MCPH1は分子のN末端部と中央部でそれぞれコンデンシンIIのHEATサブユニットCAP-D3とCAP-G2と相互作用する73).MCPH1による抑制解除がコンデンシンIIの活性化タイミングを制御することから,MCPH1とコンデンシンIIとの結合とM期特異的なリン酸化との関連性が予見される.
コンデンシンの重要性は個体レベルでも検討され,細胞やin vitroで示されてきたとおり,コンデンシンは生体でも染色体凝縮に不可欠な役割を担っている74, 75).コンデンシンを神経幹細胞において条件的にノックアウトすると,染色体の構築が乱れて,姉妹染色分体の形成不全から染色体の分配が阻害された.その結果,神経細胞の数が減少し,大脳皮質の低形成に至ることが示唆されている74).
注目すべきことに,個体レベルの解析では,コンデンシンIよりもコンデンシンIIの欠損の方が,染色体構築に大きな影響が現れる74, 75).そこでCAP-H2に点変異を持ち低機能のコンデンシンIIをかかえるマウスを解析したところ,神経幹細胞の染色体分配異常と増殖障害を伴い,小頭症様の病像を呈することが示された76).これらの結果は,コンデンシンの機能不全が小頭症の病態形成に寄与することを示唆している.実際に,小頭症患者のゲノム解析から,コンデンシンのサブユニットのCAP-D2, CAP-H, CAP-D3の変異が見つかっている.
2)がん
染色体分配の異常により染色体構造が変化する「染色体不安定性」は,多くのがんに共通して現れる性質であり,がんがゲノム的に不均一な細胞集団となることを促進する背景として重要である.染色体構造の異常は,ラギング染色体やDNAブリッジといった分配時の異常が原因で発生するので,コンデンシンをはじめとした染色体構築因子の機能不全とがんの関連が注目されている.その一方で,がんのゲノム解析によって,染色体制御因子の各ゲノム変異の頻度はそれほど高くないことがわかってきている.
コンデンシンの機能異常はがんの病態形成に寄与するのであろうか.実験的にミスセンス変異を有するCAP-H2を発現したマウスでは,染色体分配時に姉妹染色分体の絡まりの解消が遅れる.その結果,染色体構造異常や多倍体細胞を生じるが,そこにがん抑制遺伝子p53の欠失が加わり,染色体構造異常を持つ細胞の生存が許容できるようになると,T細胞性のリンパ腫が生じることが示されている77).コンデンシンとがんとの関係を探究する重要な手がかりである.
コンデンシンIとコンデンシンIIは,生物種や細胞の種類に応じて細胞内の量比はさまざまで,一口に染色体といってもその構築は多様であることが推察される.コンデンシンは五量体を形成して働き,かつこれまで述べてきたように,それぞれのサブユニットの特異的な役割が明らかになりつつある.がん細胞ではコンデンシンIとIIの量比,あるいは各サブユニットの量的な不均衡があり,病理的な染色体構造を作っている可能性があり,これからの解析を待ちたい.The Cancer Genome Atlas(TCG A)のデータによれば,コンデンシンやコヒーシンを構成するサブユニットごとの変異頻度で捉えるのではなく,一つの機能体をなす複合体として変異頻度を数えると,変異はさほど少なくはないとの指摘もあり,各サブユニットの質的変化の寄与もありそうだ78).
引用文献References
1) Paweletz, N. (2001) Nat. Rev. Mol. Cell Biol., 1, 72–75.
2) Flemming, W.(1882) Zellsubstanz, Kern und Zelltheilung, F.C.W. Vogel. Leipzig.
3) Watson, J.D. & Crick, F.H.C. (1953) Nature, 171, 737–738.
4) Kornberg, R.D. (1974) Science, 184, 868–871.
5) Sedat, J. & Manuelidis, L. (1978) Cold Spring Harb. Symp. Quant. Biol., 42, 331–350.
6) Nishino, Y., Eltsov, M., Joti, Y., Ito, K., Takata, H., Takahashi, Y., Hihara, S., Frangakis, A.S., Imamoto, N., Ishikawa, T., & Maeshima, K. (2012) EMBO J., 31, 1644–1653.
7) Ou, H.D., Phan, S., Deerinck, T.J., Thor, A., Ellisman, M.H., & O’Shea, C.C. (2017) Science, 357, eaag0025.
8) Paulson, J.R. & Laemmli, U.K. (1977) Cell, 12, 817–828.
9) Poirier, M.G. & Marko, J.F. (2002) Proc. Natl. Acad. Sci. USA, 99, 15393–15397.
10) Daban, J.R. (2015) Sci. Rep., 5, 14891.
11) Niki, H., Jaffé, A., Imamura, R., Ogura, T., & Hiraga, S. (1991) EMBO J., 10, 183–193.
12) Saitoh, N., Goldberg, I.G., Wood, E.R., & Earnshaw, W.C. (1994) J. Cell Biol., 127, 303–318.
13) Hirano, T. & Mitchison, T.J. (1994) Cell, 79, 449–458.
14) Hirano, T., Kobayashi, R., & Hirano, M. (1997) Cell, 89, 511–521.
15) Hirano, T. (2016) Cell, 164, 847–857.
16) Palecek, J.J. & Gruber, S. (2015) Structure, 23, 2183–2190.
17) Bürmann, F., Shin, H.C., Basquin, J., Soh, Y.M., Giménez-Oya, V., Kim, Y.G., Oh, B.H., & Gruber, S. (2013) Nat. Struct. Mol. Biol., 20, 371–379.
18) Ono, T., Losada, A., Hirano, M., Myers, M.P., Neuwald, A.F., & Hirano, T. (2003) Cell, 115, 109–121.
19) Hirota, T., Gerlich, D., Koch, B., Ellenberg, J., & Peters, J.M. (2004) J. Cell Sci., 117, 6435–6445.
20) Ono, T., Fang, Y., Spector, D.L., & Hirano, T. (2004) Mol. Biol. Cell, 15, 3296–3308.
21) Ono, T., Yamashita, D., & Hirano, T. (2013) J. Cell Biol., 200, 429–441.
22) Wang, B.D., Eyre, D., Basrai, M., Lichten, M., & Strunnikov, A. (2005) Mol. Cell. Biol., 25, 7216–7225.
23) D’Ambrosio, C., Schmidt, C.K., Katou, Y., Kelly, G., Itoh, T., Shirahige, K., & Uhlmann, F. (2008) Genes Dev., 22, 2215–2227.
24) Nakazawa, N., Nakamura, T., Kokubu, A., Ebe, M., Nagao, K., & Yanagida, M. (2008) J. Cell Biol., 180, 1115–1131.
25) Sutani, T., Sakata, T., Nakato, R., Masuda, K., Ishibashi, M., Yamashita, D., Suzuki, Y., Hirano, T., Bando, M., & Shirahige, K. (2015) Nat. Commun., 6, 7815.
26) Kim, K.D., Tanizawa, H., Iwasaki, O., & Noma, K. (2016) Nat. Genet., 48, 1242–1252.
27) Kranz, A.L., Jiao, C.Y., Winterkorn, L.H., Albritton, S.E., Kramer, M., & Ercan, S. (2013) Genome Biol., 14, R112.
28) Akai, Y., Kurokawa, Y., Nakazawa, N., Tonami-Murakami, Y., Suzuki, Y., Yoshimura, S.H., Iwasaki, H., Shiroiwa, Y., Nakamura, T., Shibata, E., & Yanagida, M. (2011) Open Biol., 1, 110023.
29) Wilhelm, L., Bürmann, F., Minnen, A., Shin, H.C., Toseland, C.P., Oh, B.H., & Gruber, S. (2015) eLife, 4, 06659.
30) Cuylen, S., Metz, J., & Haering, C.H. (2011) Nat. Struct. Mol. Biol., 18, 894–901.
31) Hirano, M. & Hirano, T. (2002) EMBO J., 21, 5733–5744.
32) Griese, J.J., Witte, G., & Hopfner, K.P. (2010) Nucleic Acids Res., 38, 3454–3465.
33) Hirano, M. & Hirano, T. (1998) EMBO J., 17, 7139–7148.
34) Uchiyama, S., Kawahara, K., Hosokawa, Y., Fukakusa, S., Oki, H., Nakamura, S., Kojima, Y., Noda, M., Takino, R., Miyahara, Y., Maruno, T., Kobayashi, Y., Ohkubo, T., & Fukui, K. (2015) J. Biol. Chem., 290, 29461–29477.
35) Piazza, I., Rutkowska, A., Ori, A., Walczak, M., Metz, J., Pelechano, V., Beck, M., & Haering, C.H. (2014) Nat. Struct. Mol. Biol., 21, 560–568.
36) Soh, Y.M., Bürmann, F., Shin, H.C., Oda, T., Jin, K.S., Toseland, C.P., Kim, C., Lee, H., Kim, S.J., Kong, M.S., Durand-Diebold, M.L., Kim, Y.G., Kim, H.M., Lee, N.K., Sato, M., Oh, B.H., & Gruber, S. (2015) Mol. Cell, 57, 290–303.
37) Kimura, K., Hirano, M., Kobayashi, R., & Hirano, T. (1998) Science, 282, 487–490.
38) Abe, S., Nagasaka, K., Hirayama, Y., Kozuka-Hata, H., Oyama, M., Aoyagi, Y., Obuse, C., & Hirota, T. (2011) Genes Dev., 25, 863–874.
39) Robellet, X., Thattikota, Y., Wang, F., Wee, T.L., Pascariu, M., Shankar, S., Bonneil, É., Brown, C.M., & D’Amours, D. (2015) Genes Dev., 29, 426–439.
40) Sutani, T., Yuasa, T., Tomonaga, T., Dohmae, N., Takio, K., & Yanagida, M. (1999) Genes Dev., 13, 2271–2283.
41) St-Pierre, J., Douziech, M., Bazile, F., Pascariu, M., Bonneil, E., Sauvé, V., Ratsima, H., & D’Amours, D. (2009) Mol. Cell, 34, 416–426.
42) Lipp, J.J., Hirota, T., Poser, I., & Peters, J.M. (2007) J. Cell Sci., 120, 1245–1255.
43) Takemoto, A., Murayama, A., Katano, M., Urano, T., Furukawa, K., Yokoyama, S., Yanagisawa, J., Hanaoka, F., & Kimura, K. (2007) Nucleic Acids Res., 35, 2403–2412.
44) Lavoie, B.D., Hogan, E., & Koshland, D. (2004) Genes Dev., 18, 76–87.
45) Mora-Bermúdez, F., Gerlich, D., & Ellenberg, J. (2007) Nat. Cell Biol., 9, 822–831.
46) Takemoto, A., Kimura, K., Yanagisawa, J., Yokoyama, S., & Hanaoka, F. (2006) EMBO J., 25, 5339–5348.
47) Rusin, S.F., Schlosser, K.A., Adamo, M.E., & Kettenbach, A.N. (2015) Sci. Signal., 8, rs12.
48) Maeshima, K. & Laemmli, U.K. (2003) Dev. Cell, 4, 467–480.
49) Savvidou, E., Cobbe, N., Steffensen, S., Cotterill, S., & Heck, M.M. (2005) J. Cell Sci., 118, 2529–2543.
50) Baxter, J., Sen, N., Martínez, V.L., De Carandini, M.E., Schvartzman, J.B., Diffley, J.F., & Aragón, L. (2011) Science, 331, 1328–1332.
51) Charbin, A., Bouchoux, C., & Uhlmann, F. (2014) Nucleic Acids Res., 42, 340–348.
52) Nagasaka, K., Hossain, M.J., Roberti, M.J., Ellenberg, J., & Hirota, T. (2016) Nat. Cell Biol., 18, 692–699.
53) Sen, N., Leonard, J., Torres, R., Garcia-Luis, J., Palou-Marin, G., & Aragón, L. (2016) Mol. Cell, 64, 134–147.
54) Bhat, M.A., Philp, A.V., Glover, D.M., & Bellen, H.J. (1996) Cell, 87, 1103–1114.
55) Hayama, R. & Marians, K.J. (2010) Proc. Natl. Acad. Sci. USA, 107, 18826–18831.
56) Li, Y., Stewart, N.K., Berger, A.J., Vos, S., Schoeffler, A.J., Berger, J.M., Chait, B.T., & Oakley, M.G. (2010) Proc. Natl. Acad. Sci. USA, 107, 18832–18837.
57) Takahashi, M., Tanaka, K., Wakai, T., & Hirota, T. (2016) Biomed. Res., 37, 161–165.
58) Takahashi, M., Wakai, T., & Hirota, T. (2016) Genes Dev., 30, 1931–1936.
59) Wilkins, B.J., Rall, N.A., Ostwal, Y., Kruitwagen, T., Hiragami-Hamada, K., Winkler, M., Barral, Y., Fischle, W., & Neumann, H. (2014) Science, 343, 77–80.
60) Vagnarelli, P., Hudson, D.F., Ribeiro, S.A., Trinkle-Mulcahy, L., Spence, J.M., Lai, F., Farr, C.J., Lamond, A.I., & Earnshaw, W.C. (2006) Nat. Cell Biol., 8, 1133–1142.
61) Booth, D.G., Beckett, A.J., Molina, O., Samejima, I., Masumoto, H., Kouprina, N., Larionov, V., Prior, I.A., & Earnshaw, W.C. (2016) Mol. Cell, 64, 790–802.
62) Verheijen, R., Kuijpers, H.J., van Driel, R., Beck, J.L., van Dierendonck, J.H., Brakenhoff, G.J., & Ramaekers, F.C. (1989) J. Cell Sci., 92, 531–540.
63) Cuylen, S., Blaukopf, C., Politi, A.Z., Müller-Reichert, T., Neumann, B., Poser, I., Ellenberg, J., Hyman, A.A., & Gerlich, D.W. (2016) Nature, 535, 308–312.
64) Booth, D.G., Takagi, M., Sanchez-Pulido, L., Petfalski, E., Vargiu, G., Samejima, K., Imamoto, N., Ponting, C.P., Tollervey, D., Earnshaw, W.C., & Vagnarelli, P. (2014) eLife, 3, e01641.
65) Gerlich, D., Hirota, T., Koch, B., Peters, J.M., & Ellenberg, J. (2006) Curr. Biol., 16, 333–344.
66) Liang, Z., Zickler, D., Prentiss, M., Chang, F.S., Witz, G., Maeshima, K., & Kleckner, N. (2015) Cell, 161, 1124–1137.
67) Petrova, B., Dehler, S., Kruitwagen, T., Hériché, J.K., Miura, K., & Haering, C.H. (2013) Mol. Cell. Biol., 33, 984–998.
68) Hériché, J.K., Lees, J.G., Morilla, I., Walter, T., Petrova, B., Roberti, M.J., Hossain, M.J., Adler, P., Fernández, J.M., Krallinger, M., Haering, C.H., Vilo, J., Valencia, A., Ranea, J.A., Orengo, C., & Ellenberg, J. (2014) Mol. Biol. Cell, 25, 2522–2536.
69) Poonperm, R., Takata, H., Hamano, T., Matsuda, A., Uchiyama, S., Hiraoka, Y., & Fukui, K. (2015) Sci. Rep., 5, 11916.
70) Markaki, Y., Gunkel, M., Schermelleh, L., Beichmanis, S., Neumann, J., Heidemann, M., Leonhardt, H., Eick, D., Cremer, C., & Cremer, T. (2010) Cold Spring Harb. Symp. Quant. Biol., 75, 475–492.
71) Naumova, N., Imakaev, M., Fudenberg, G., Zhan, Y., Lajoie, B.R., Mirny, L.A., & Dekker, J. (2013) Science, 342, 948–953.
72) Trimborn, M., Schindler, D., Neitzel, H., & Hirano, T. (2006) Cell Cycle, 5, 322–326.
73) Yamashita, D., Shintomi, K., Ono, T., Gavvovidis, I., Schindler, D., Neitzel, H., Trimborn, M., & Hirano, T. (2011) J. Cell Biol., 194, 841–854.
74) Nishide, K. & Hirano, T. (2014) PLoS Genet., 10, e1004847.
75) Houlard, M., Godwin, J., Metson, J., Lee, J., Hirano, T., & Nasmyth, K. (2015) Nat. Cell Biol., 17, 771–781.
76) Martin, C.A., Murray, J.E., Carroll, P., Leitch, A., Mackenzie, K.J., Halachev, M., Fetit, A.E., Keith, C., Bicknell, L.S., Fluteau, A., Gautier, P., Hall, E.A., Joss, S., Soares, G., Silva, J., Bober, M.B., Duker, A., Wise, C.A., Quigley, A.J., Phadke, S.R., Wood, A.J., Vagnarelli, P., & Jackson, A.P.; Deciphering Developmental Disorders Study. (2016) Genes Dev., 30, 2158–2172.
77) Woodward, J., Taylor, G.C., Soares, D.C., Boyle, S., Sie, D., Read, D., Chathoth, K., Vukovic, M., Tarrats, N., Jamieson, D., Campbell, K.J., Blyth, K., Acosta, J.C., Ylstra, B., Arends, M.J., Kranc, K.R., Jackson, A.P., Bickmore, W.A., & Wood, A.J. (2016) Genes Dev., 30, 2173–2186.
78) Leiserson, M.D., Vandin, F., Wu, H.T., Dobson, J.R., Eldridge, J.V., Thomas, J.L., Papoutsaki, A., Kim, Y., Niu, B., McLellan, M., Lawrence, M.S., Gonzalez-Perez, A., Tamborero, D., Cheng, Y., Ryslik, G.A., Lopez-Bigas, N., Getz, G., Ding, L., & Raphael, B.J. (2015) Nat. Genet., 47, 106–114.