NAD+代謝・サーチュインと幹細胞老化NAD+ metabolism and Sirtuin in stem cell aging
東京大学大学院医学系研究科 代謝栄養病態学Department of Diabetes and Metabolic Diseases, The University of Tokyo ◇ 東京都文京区本郷7–3–1 ◇ 7–3–1 Hongo Bunkyo-ku Tokyo
発行日:2017年8月25日Published: August 25, 2017
© 2017 公益社団法人日本生化学会© 2017 The Japanese Biochemical Society
老化は,がん,糖尿病,心血管疾患などの成人病やアルツハイマー病,パーキンソン病などの神経疾患の危険因子の一つである.各臓器,組織の恒常性は,それぞれの組織の幹細胞によって維持されている.組織幹細胞の多くは老化によりその機能が低下することが示されており,老化関連疾患の原因となりうる.近年,老化および老化関連疾患は,ニコチンアミドアデニンジヌクレオチド(nicotinamide adenine dinucleotide:NAD+)量低下,およびNAD+依存性脱アセチル化酵素サーチュインの活性低下と密接な関わりを持つことが示されている1, 2).また,サーチュインの活性化は,カロリー制限における寿命延長や健康増進に関わる効果の多くを説明すると考えられている2).本稿では,NAD+代謝とサーチュイン活性の観点から,老化,およびカロリー制限における幹細胞制御の最新の知見を概説する.
NAD+は,古くは,酸化還元反応の補酵素として知られるが,近年は,ポリADPリボースポリメラーゼ(poly(ADP ribose)polymerases:PARPs),CD38/CD157やサーチュインの基質としての役割が知られるようになった1).特に,サーチュインによるNAD+からニコチンアミド(nicotinamide:NAM)への分解反応は,それと共役するサーチュインによるリシン残基脱アセチル化反応を促進することで,健康,長寿に関わるさまざまな生命現象に関与している1, 2).哺乳類では,七つのサーチュイン(SIRT1~7)が存在し,SIRT1, 6, 7は核内,SIRT3, 4, 5はミトコンドリア,SIRT2は細胞質に局在する2).最も研究が進んでいるSIRT1はNAD+依存的に,時計遺伝子(BMAL1, CLOCK)を制御するが,逆に,NAMからNAD+を再合成する律速酵素ニコチンアミドホスホリボシルトランスフェラーゼ(nicotinamide phosphoribosyltransferase:NAMPT)は,時計遺伝子によって制御されており,NAD+代謝は慨日リズムを刻んでいる1)(図1).老化に伴いNAD+量およびサーチュイン活性が低下するが1),NAMPTの酵素反応産物であるニコチンアミドモノヌクレオチド(nicotinamide mononucleotide:NMN)や,ニコチンアミドリボシド(nicotinamide riboside:NR)などのNAD+中間代謝産物の補充がサーチュインを効果的に再活性化する1).また,NAD+量は栄養状態により変化するが,カロリー制限におけるNAD+増加は,カロリー制限による寿命延長や健康増進の効果を媒介すると考えられている1).
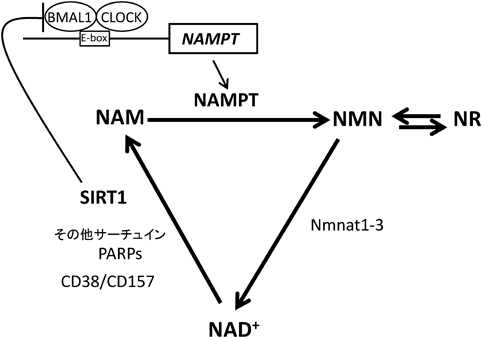
NAMPT(nicotinamide phosphoribosyltransferase)はNAD+合成系の律速酵素として働き,NAM(nicotinamide)からNMN(nicotinamide mononucleotide)を産生する.NAD+は,SIRT1を含むサーチュイン,PARPs [poly(ADP ribose)polymerases], CD38/CD157などが媒介する酵素反応の基質として利用される.また,NAD+は,SIRT1を介してCLOCK:BMAL1複合体によるNAMPT転写活性化を抑制し,負のフィードバックループを形成している.Nmnat:nicotinamide mononucleotide adenylyltransferase, NR:nicotinamide riboside.
組織の修復,代謝回転は,正常な組織恒常性の維持に必要不可欠な過程であり,組織幹細胞が重要な役割を果たす.一般に,老化により,幹細胞は自己複製および分化能力を失い,組織の退化につながる.カロリー制限は,多くの生物種において寿命延長効果および健康促進効果を持つことが示されているが,老化により低下する幹細胞機能を維持する介入手段でもある3).カロリー制限の効果は,いくつかの経路が媒介することが知られている.1)ホスファチジルイノシトール3-キナーゼ(phosphatidylinositol 3-kinase:PI3K)/AKT経路の低下4),2)mechanistic target of rapamycin(mTOR)活性の低下4),3)AMP活性化プロテインキナーゼ(AMP-activated protein kinase:AMPK)活性化2, 4),4)NAD+上昇を通じたサーチュイン活性化1, 2)が主要な経路として知られるが,これらを通じて,幹細胞を保護するようなストレス応答経路を活性化するものと考えられる3).
小腸では,幹細胞ニッチであるパネート細胞と腸管上皮幹細胞が隣接しており,ニッチと幹細胞との相互作用を調べる上で,理想的な臓器といえる.ここでは,カロリー制限における腸管上皮幹細胞ニッチ(パネート細胞)と腸管上皮幹細胞の制御について得られている最新の知見について紹介する.
腸管上皮幹細胞は,そのニッチ細胞であるパネート細胞から分泌されるWntなどの液性因子によりその自己複製能が制御されている5).カロリー制限によって,腸管上皮幹細胞はその数と自己複製能を増加させるが,その増加は,パネート細胞から分泌される環状アデノシン二リン酸リボース(cyclic ADP-ribose:cADPR)によることが報告された6).カロリー制限は,パネート細胞において,mTOR複合体1(mTORC1)活性の低下を通じて,CD157活性を上昇させる.CD157によるNAD+の分解によって生成されたcADPRがパネート細胞から分泌されて,幹細胞の自己複製を誘導する(図2)6).
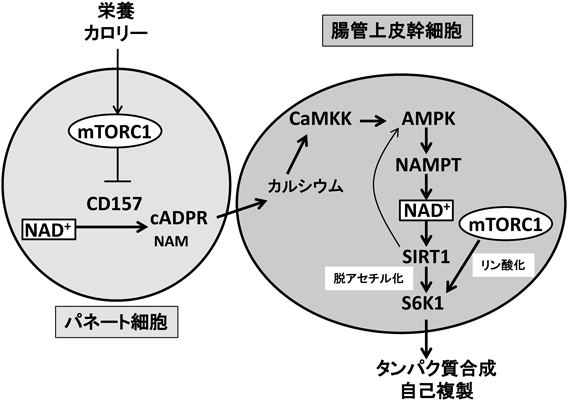
カロリー制限は,パネート細胞において,mTOR複合体1(mTORC1)の低下を介し,CD157を活性化して,NAD+からのcADPR(cyclic ADP-ribose)生成を促進する.パネート細胞より分泌されたcADPRは,細胞内カルシウムの上昇,CaMKK(calcium-calmodulin-dependent protein kinase kinase)の活性化,AMPK (AMP-activated protein kinase)/NAMPTの活性化を介して,腸管上皮幹細胞内のNAD+量とSIRT1活性を増加させる.SIRT1は,p70-S6キナーゼ1(S6K1)を脱アセチル化し,mTORC1によるS6K1リン酸化を増加させる.パネート細胞と腸管上皮幹細胞の両細胞において,NAD+の上昇は重要な役割を持つ.一方,カロリー制限に関して,mTORC1は両細胞において重要な役割を持つが,腸管上皮幹細胞でのmTORC1の変化は,パネート細胞での変化の正反対である.
最近,著者らは,思いがけないことに,腸管上皮幹細胞におけるmTORC1活性がパネート細胞および腸絨毛細胞におけるmTORC1活性とは異なって上昇しており,その結果,腸管上皮幹細胞におけるタンパク質合成および自己複製能が増加することを見いだした7, 8).また,遺伝子改変マウスを解析し,パネート細胞と腸管上皮幹細胞の共培養系を確立することで,カロリー制限における腸管上皮幹細胞自己複製のシグナル経路を明らかにした7).具体的には,cADPRは1)細胞内カルシウム上昇,2)カルシウム・カルモジュリン依存性プロテインキナーゼキナーゼ(calcium-calmodulin-dependent protein kinase kinase:CaMKK)活性上昇によるAMPK活性化,3)AMPK活性化によるNAD+合成酵素NAMPTの発現上昇9),4)NAD+上昇によるSIRT1の活性化,5)SIRT1によるS6K1脱アセチル化を通じた,mTORC1活性化によるp70-S6キナーゼ1(S6K1)リン酸化の上昇10)を引き起こす.これらのシグナル伝達経路が,カロリー制限下の腸管上皮幹細胞でのmTORC1シグナル増加を可能にしている(図2).
腸管上皮幹細胞でのSIRT1ノックダウンにより,カロリー制限下パネート細胞からのシグナルへの腸管上皮幹細胞の応答は消失するが,パネート細胞でのSIRT1ノックダウンはカロリー制限の効果に影響を与えない7).また,自由摂食下でSIRT1トランスジェニックマウスは,カロリー制限下マウスと同様に,腸管上皮幹細胞自己複製の増加,腸絨毛の短縮を認め,腸管上皮幹細胞でのSIRT1活性化は,SIRT1/mTORC1シグナル活性化に必要十分であると考えられる7).
一般的にカロリー制限において,mTORC1活性は,PI3K/AKTシグナルの低下あるいはAMPK活性化により抑制されうるが4),カロリー制限下腸管上皮幹細胞においてこれらの変化は認められない7).腸管上皮幹細胞は,カロリー制限による直接の栄養,エネルギー状態の感知機構を兼ね備えておらず,パネート細胞から分泌されるcADPRを通じたSIRT1活性の変化にのみ制御されていると考えられる.
mTORインヒビターであるラパマイシンの投与は,オートファジーの亢進などを通じ,寿命延長効果を含めたカロリー制限の多くの効果を模倣するといわれている4).しかし,このカロリー制限下腸管上皮幹細胞において新しく同定された経路(図2)から推測されるように,ラパマイシンは,カロリー制限による腸管上皮幹細胞自己複製の増加を抑制し,また,自由摂食下の腸管上皮幹細胞の自己複製を増加させない7).このように,カロリー制限において,パネート細胞と腸管上皮幹細胞におけるmTORC1活性の変化が正反対であることから,ラパマイシンを腸管上皮幹細胞におけるカロリー制限の効果を模倣する薬剤として使用することは困難であるといえる.一方,SIRT1活性化薬剤やNMNおよびNRなどのNAD+中間代謝産物の投与は,カロリー制限同様に,効果的に腸管上皮幹細胞の自己複製を増加させる可能性がある.特に,NAD+の補充はパネート細胞におけるcADPR合成を促進し6),かつ腸管上皮幹細胞のSIRT1を活性化させることから7),NRあるいはNMNによるNAD+補充が,老化における腸管上皮幹細胞の機能低下を回復させる有効な手段となる可能性を示している.
これまで述べたように,老化により幹細胞は自己複製能力を失い,その機能低下が老化による組織退化につながる3).老化に伴うNAD+量の低下,およびそれに伴うサーチュイン(SIRT1~7)活性低下が幹細胞の機能低下の原因となりうるが,NAD+量やサーチュイン活性を回復することは,老化による幹細胞機能低下を防ぐ有効な手段となりうる.ここに,幹細胞老化とNAD+およびサーチュインに関わるいくつかの報告について概説する.
ミトコンドリアタンパク質のアセチル化を制御し,酸化ストレスを軽減する役割を持つSIRT3は,血液幹細胞に多くの発現を認め,ストレス条件下および老化における,血液幹細胞と組織恒常性の維持において重要な役割を持つ11).SIRT3の発現は老齢マウスの血液幹細胞において抑制されているが,SIRT3の過剰発現は,老齢マウスの血液幹細胞の自己複製能を回復し,SIRT3がミトコンドリア代謝を通じて,血液幹細胞老化を制御することが示されている11).
また,別の報告では,SIRT7とnuclear respiratory factor 1(NRF1)がミトコンドリアの異常タンパク質応答(mitochondrial unfolded protein response:UPRmt)を通じ,血液幹細胞の自己複製能を制御することが示されている12).SIRT7の不活性化は,UPRmtの増加と,幹細胞自己複製能の低下を引き起こす.一方,SIRT7の発現量は,老齢マウスの血液幹細胞で減少しており,SIRT7の過剰発現は,老齢マウスの血液幹細胞における自己複製能を改善する12).以上の報告は,ミトコンドリア代謝が血液幹細胞老化において重要であるが,サーチュイン活性化は,老化におけるミトコンドリア機能の低下を改善することを示している.
老齢マウスでは,骨格筋繊維の前駆細胞である筋衛星細胞におけるミトコンドリア遺伝子の発現が低下している13).老齢マウスへのNAD+中間代謝産物NRの投与は,筋衛星細胞のミトコンドリア機能,および自己複製能と筋繊維への分化能を改善する13).また,NRの投与は,老齢マウスの運動能力,筋への化学物質によるダメージに対する修復能を改善し,老齢マウスの寿命を延長させる13).これらNRの幹細胞機能改善の効果は,筋肉に限らず,神経幹細胞など他の組織幹細胞でも認められる13).NAD+の上昇が,SIRT1依存的に,UPRmtのメディエーターであるプロヒビチン(prohibitin:PHB)を活性化することで,老齢マウスの幹細胞におけるUPRmtの機能を改善することが示されている13).
近年,老化において低下したNAD+量やサーチュイン活性を回復することは,さまざまな老化関連疾患への有効なアプローチであることが動物実験によって示されている14).本稿では,NAD+量やサーチュイン活性の回復が,老化において低下するさまざまな組織幹細胞の機能を回復する有効な手段であることを示す最新の知見を紹介した.今後,これらのアプローチのヒトへの応用が期待されるところであり,臨床試験の結果が待たれる.
留学にてご指導いただいたマサチューセッツ工科大学生物学部のLeonard Guarente教授にこの場を借りて厚く御礼申し上げます.また,著者の留学に際してご支援をいただいた鈴木万平糖尿病財団に厚く御礼申し上げます.
1) Imai, S. & Guarente, L. (2014) Trends Cell Biol., 24, 464–471.
2) Guarente, L. (2013) Genes Dev., 27, 2072–2085.
3) Schultz, M.B. & Sinclair, D.A. (2016) Development, 143, 3–14.
4) Johnson, S.C., Rabinovitch, P.S., & Kaeberlein, M. (2013) Nature, 493, 338–345.
5) Sato, T., van Es, J.H., Snippert, H.J., Stange, D.E., Vries, R.G., van den Born, M., Barker, N., Shroyer, N.F., van de Wetering, M., & Clevers, H. (2011) Nature, 469, 415–418.
6) Yilmaz, Ö.H., Katajisto, P., Lamming, D.W., Gültekin, Y., Bauer-Rowe, K.E., Sengupta, S., Birsoy, K., Dursun, A., Yilmaz, V.O., Selig, M., Nielsen, G.P., Mino-Kenudson, M., Zukerberg, L.R., Bhan, A.K., Deshpande, V., & Sabatini, D.M. (2012) Nature, 486, 490–495.
7) Igarashi, M. & Guarente, L. (2016) Cell, 166, 436–450.
8) Igarashi, M. & Guarente, L. (2016) Cell Cycle, 16, 1–2.
9) Cantó, C., Gerhart-Hines, Z., Feige, J.N., Lagouge, M., Noriega, L., Milne, J.C., Elliott, P.J., Puigserver, P., & Auwerx, J. (2009) Nature, 458, 1056–1060.
10) Hong, S., Zhao, B., Lombard, D.B., Fingar, D.C., & Inoki, K. (2014) J. Biol. Chem., 289, 13132–13141.
11) Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., Zhang, D., Scadden, D.T., & Chen, D. (2013) Cell Reports, 3, 319–327.
12) Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., Haynes, C.M., & Chen, D. (2015) Science, 347, 1374–1377.
13) Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., D’Amico, D., Ropelle, E.R., Lutolf, M.P., Aebersold, R., Schoonjans, K., Menzies, K.J., & Auwerx, J. (2016) Science, 352, 1436–1443.
14) Mills, K.F., Yoshida, S., Stein, L.R., Grozio, A., Kubota, S., Sasaki, Y., Redpath, P., Migaud, M.E., Apte, R.S., Uchida, K., Yoshino, J., & Imai, S.I. (2016) Cell Metab., 24, 795–806.

東京大学大学院医学系研究科 代謝栄養病態学助教.東京大学大学院内科学専攻博士課程修了.
1976年東京都に生る.2002年東京大学医学部卒業.08年同大学院内科学専攻博士課程修了.10年マサチューセッツ工科大学生物学部研究員.17年より現職.
研究テーマと抱負食事による幹細胞の制御を軸に,老化による組織の機能低下や,発癌の過程のメカニズムを理解し,それをもとに,組織の若返りや癌の治療を考察していきたい.
趣味野球観戦.

This page was created on 2017-06-27T13:30:19.339+09:00
This page was last modified on 2017-08-18T09:26:28.433+09:00
このサイトは(株)国際文献社によって運用されています。