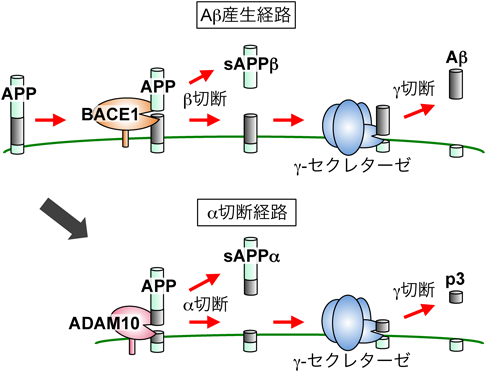
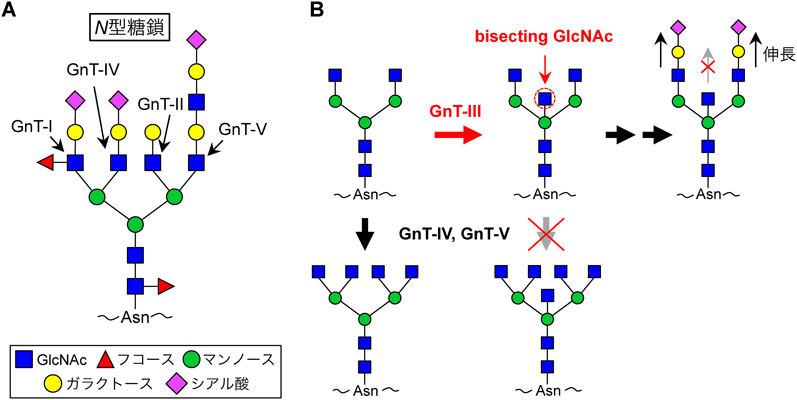
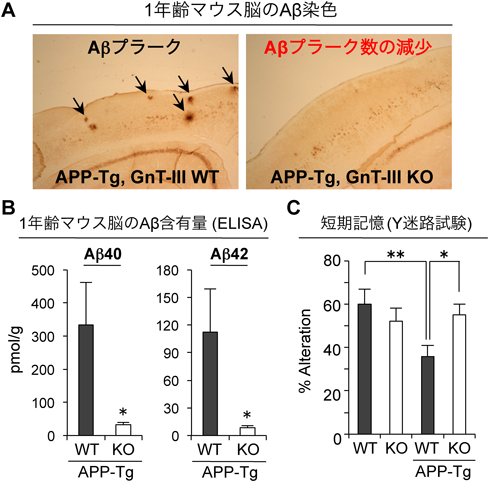
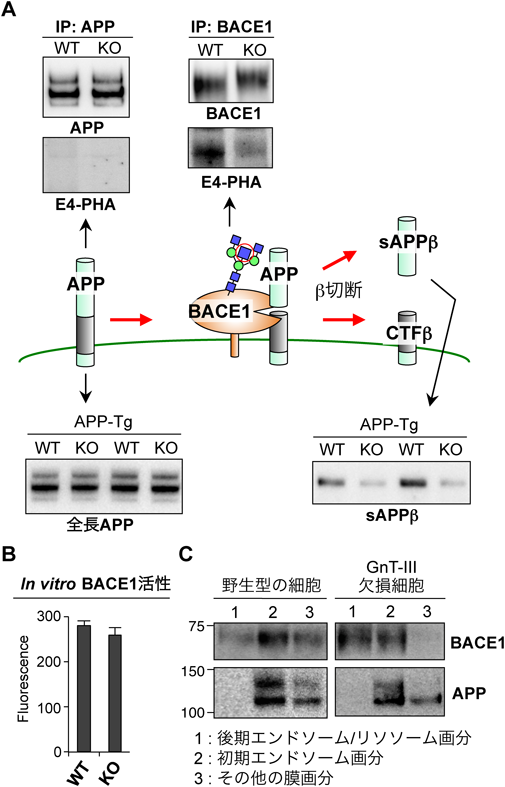
1) Scheltens, P., Blennow, K., Breteler, M.M., de Strooper, B., Frisoni, G.B., Salloway, S., & Van der Flier, W.M. (2016) Lancet, 388, 505–517.
3) Porteri, C., Albanese, E., Scerri, C., Carrillo, M.C., Snyder, H.M., Martensson, B., Baker, M., Giacobini, E., Boccardi, M., Winblad, B., Frisoni, G.B., & Hurst, S.; Geneva Task Force for the Roadmap of Alzheimer’s Biomarkers (2017) Neurobiol. Aging, 52, 132–140.
5) Rogaev, E.I., Sherrington, R., Rogaeva, E.A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., Mar, L., Sorbi, S., Nacmias, B., Piacentini, S., Amaducci, L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P.E., Rommens, J.M., & George-Hyslop, P.H.S. (1995) Nature, 376, 775–778.
6) Sherrington, R., Rogaev, E.I., Liang, Y., Rogaeva, E.A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J.F., Bruni, A.C., Montesi, M.P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau, P., Polinsky, R.J., Wasco, W., Da Silva, H.A., Haines, J.L., Perkicak-Vance, M.A., Tanzi, R.E., Roses, A.D., Fraser, P.E., Rommens, J.M., & St George-Hyslop, P.H. (1995) Nature, 375, 754–760.
9) Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., & Lannfelt, L. (1992) Nat. Genet., 1, 345–347.
13) Kizuka, Y., Kitazume, S., & Taniguchi, N. (2017) Biochim. Biophys. Acta. 10.1016/j.bbagen.2017.04.012
14) Stanley, P., Schachter, H., & Taniguchi, N.(2009) N-Glycans. in Essentials of Glycobiology (Varki, A., Cummings, R. D., Esko, J. D., Freeze, H. H., Stanley, P., Bertozzi, C. R., Hart, G. W., & Etzler, M. E. eds.), 2nd Ed., Cold Spring Harbor (NY). pp 101–114.
16) Granovsky, M., Fata, J., Pawling, J., Muller, W.J., Khokha, R., & Dennis, J.W. (2000) Nat. Med., 6, 306–312.
18) Nishikawa, A., Ihara, Y., Hatakeyama, M., Kangawa, K., & Taniguchi, N. (1992) J. Biol. Chem., 267, 18199–18204.
20) Gu, J., Nishikawa, A., Tsuruoka, N., Ohno, M., Yamaguchi, N., Kangawa, K., & Taniguchi, N. (1993) J. Biochem., 113, 614–619.
23) Miyoshi, E., Uozumi, N., Noda, K., Hayashi, N., Hori, M., & Taniguchi, N. (1997) Int. J. Cancer, 72, 1117–1121.
25) Akasaka-Manya, K., Manya, H., Sakurai, Y., Wojczyk, B.S., Kozutsumi, Y., Saito, Y., Taniguchi, N., Murayama, S., Spitalnik, S.L., & Endo, T. (2010) Glycobiology, 20, 99–106.
27) Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., Iwata, N., & Saido, T.C. (2014) Nat. Neurosci., 17, 661–663.
28) Kizuka, Y., Kitazume, S., Fujinawa, R., Saito, T., Iwata, N., Saido, T.C., Nakano, M., Yamaguchi, Y., Hashimoto, Y., Staufenbiel, M., Hatsuta, H., Murayama, S., Manya, H., Endo, T., & Taniguchi, N. (2015) EMBO Mol. Med., 7, 175–189.
30) Nagae, M., Kanagawa, M., Morita-Matsumoto, K., Hanashima, S., Kizuka, Y., Taniguchi, N., & Yamaguchi, Y. (2016) Sci. Rep., 6, 22973.
31) Cummings, R.D. & Kornfeld, S. (1982) J. Biol. Chem., 257, 11230–11234.
32) Tan, J. & Evin, G. (2012) J. Neurochem., 120, 869–880.
33) Das, U., Scott, D.A., Ganguly, A., Koo, E.H., Tang, Y., & Roy, S. (2013) Neuron, 79, 447–460.
35) Vassar, R., Kuhn, P.H., Haass, C., Kennedy, M.E., Rajendran, L., Wong, P.C., & Lichtenthaler, S.F. (2014) J. Neurochem., 130, 4–28.
36) Yang, L.B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X.L., Beach, T., Sue, L., Wong, P., Price, D., Li, R., & Shen, Y. (2003) Nat. Med., 9, 3–4.
38) Kizuka, Y., Nakano, M., Kitazume, S., Saito, T., Saido, T.C., & Taniguchi, N. (2016) Biochem. J., 473, 21–30.
40) Herreman, A., Hartmann, D., Annaert, W., Saftig, P., Craessaerts, K., Serneels, L., Umans, L., Schrijvers, V., Checler, F., Vanderstichele, H., Baekelandt, V., Dressel, R., Cupers, P., Huylebroeck, D., Zwijsen, A., Van Leuven, F., & De Strooper, B. (1999) Proc. Natl. Acad. Sci. USA, 96, 11872–11877.
41) Donoviel, D.B., Hadjantonakis, A.K., Ikeda, M., Zheng, H., Hyslop, P.S., & Bernstein, A. (1999) Genes Dev., 13, 2801–2810.
42) Willem, M., Garratt, A.N., Novak, B., Citron, M., Kaufmann, S., Rittger, A., DeStrooper, B., Saftig, P., Birchmeier, C., & Haass, C. (2006) Science, 314, 664–666.
43) Savonenko, A.V., Melnikova, T., Laird, F.M., Stewart, K.A., Price, D.L., & Wong, P.C. (2008) Proc. Natl. Acad. Sci. USA, 105, 5585–5590.
46) Zentella, R., Sui, N., Barnhill, B., Hsieh, W.P., Hu, J., Shabanowitz, J., Boyce, M., Olszewski, N.E., Zhou, P., Hunt, D.F., & Sun, T.P. (2017) Nat. Chem. Biol., 13, 479–485.