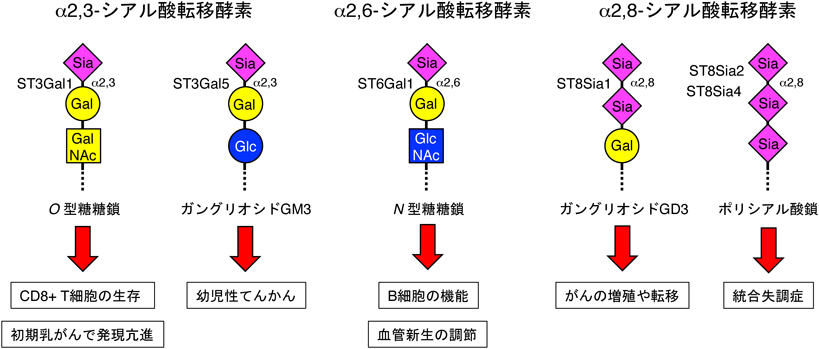
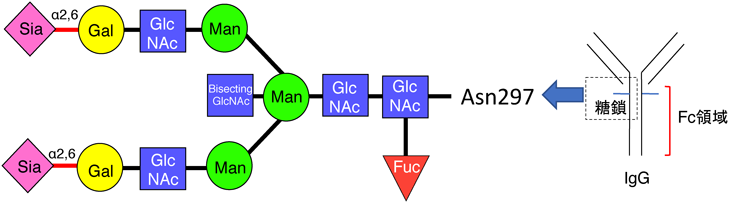
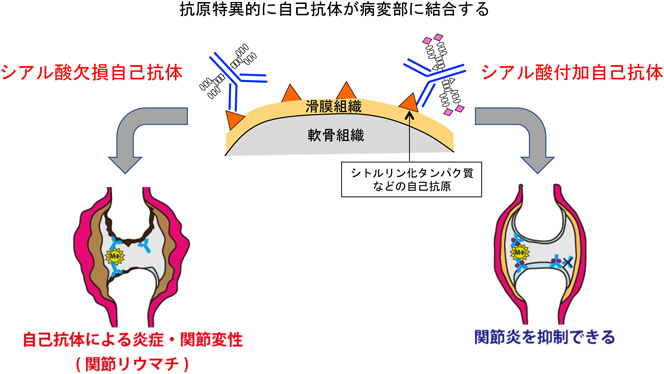
1) Priatel, J.J., Chui, D., Hiraoka, N., Simmons, C.J., Richardson, K.B., Page, D.M., Fukuda, M., Varki, N.M., & Marth, J.D. (2000) Immunity, 12, 273–283.
2) Burchell, J., Poulsom, R., Hanby, A., Whitehouse, C., Cooper, L., Clausen, H., Miles, D., & Taylor-Papadimitriou, J. (1999) Glycobiology, 9, 1307–1311.
3) Simpson, M.A., Cross, H., Proukakis, C., Priestman, D.A., Neville, D.C., Reinkensmeier, G., Wang, H., Wiznitzer, M., Gurtz, K., Verganelaki, A., Pryde, A., Patton, M.A., Dwek, R.A., Butters, T.D., Platt, F.M., & Crosby, A.H. (2004) Nat. Genet., 36, 1225–1229.
4) Hennet, T., Chui, D., Paulson, J.C., & Marth, J.D. (1998) Proc. Natl. Acad. Sci. USA, 95, 4504–4509.
5) Kitazume, S., Imamaki, R., Ogawa, K., Komi, Y., Futakawa, S., Kojima, S., Hashimoto, Y., Marth, J.D., Paulson, J.C., & Taniguchi, N. (2010) J. Biol. Chem., 285, 6515–6521.
6) Ohkawa, Y., Momota, H., Kato, A., Hashimoto, N., Tsuda, Y., Kotani, N., Honke, K., Suzumura, A., Furukawa, K., Ohmi, Y., Natsume, A., Wakabayashi, T., & Furukawa, K. (2015) J. Biol. Chem., 290, 16043–16058.
7) Sato, C. & Kitajima, K. (2013) Front. Cell. Neurosci., 7, 61.
8) Krocher, T., Rockle, I., Diederichs, U., Weinhold, B., Burkhardt, H., Yanagawa, Y., Gerardy-Schahn, R., & Hildebrandt, H. (2014) Development, 141, 3022–3032.
9) Ohmi, Y., Ise, W., Harazono, A., Takakura, D., Fukuyama, H., Baba, Y., Narazaki, M., Shoda, H., Takahashi, N., Ohkawa, Y., Ji, S., Sugiyama, F., Fujio, K., Kumanogoh, A., Yamamoto, K., Kawasaki, N., Kurosaki, T., Takahashi, Y., & Furukawa, K. (2016) Nat. Commun., 7, 11205.
11) Kuhn, K.A., Kulik, L., Tomooka, B., Braschler, K.J., Arend, W.P., Robinson, W.H., & Holers, V.M. (2006) J. Clin. Invest., 116, 961–973.
12) Cantaert, T., De Rycke, L., Bongartz, T., Matteson, E.L., Tak, P.P., Nicholas, A.P., & Baeten, D. (2006) Arthritis Rheum., 54, 3381–3389.
13) Verpoort, K.N., Jol-van der Zijde, C.M., Papendrecht-van der Voort, E.A., Ioan-Facsinay, A., Drijfhout, J.W., van Tol, M.J., Breedveld, F.C., Huizinga, T.W., & Toes, R.E. (2006) Arthritis Rheum., 54, 3799–3808.
14) Schuerwegh, A.J., Ioan-Facsinay, A., Dorjee, A.L., Roos, J., Bajema, I.M., van der Voort, E.I., Huizinga, T.W., & Toes, R.E. (2010) Proc. Natl. Acad. Sci. USA, 107, 2586–2591.
15) Suwannalai, P., Willemze, A., van Toorn, L., Stoeken-Rijsbergen, G., Levarht, N., Drijfhout, J.W., Huizinga, T.W., Toes, R.E., & Trouw, L.A. (2006) Arthritis Rheum., 54, 3799–3808.
16) Uysal, H., Bockermann, R., Nandakumar, K.S., Sehnert, B., Bajtner, E., Engstrom, A., Serre, G., Burkhardt, H., Thunnissen, M.M., & Holmdahl, R. (2009) J. Exp. Med., 206, 449–462.
18) Rudd, P.M., Leatherbarrow, R.J., Rademacher, T.W., & Dwek, R.A. (1991) Mol. Immunol., 28, 1369–1378.
19) Shinkawa, T., Nakamura, K., Yamane, N., Shoji-Hosaka, E., Kanda, Y., Sakurada, M., Uchida, K., Anazawa, H., Satoh, M., Yamasaki, M., Hanai, N., & Shitara, K. (2003) J. Biol. Chem., 278, 3466–3473.
20) Shibata-Koyama, M., Iida, S., Okazaki, A., Mori, K., Kitajima-Miyama, K., Saitou, S., Kakita, S., Kanda, Y., Shitara, K., Kato, K., & Satoh, M. (2009) Glycobiology, 19, 126–134.
21) Parekh, R.B., Dwek, R.A., Sutton, B.J., Fernandes, D.L., Leung, A., Stanworth, D., Rademacher, T.W., Mizuochi, T., Taniguchi, T., Matsuta, K., Takeuchi, F., Nagato, Y., Miyamoto, T., & Kobata, A. (1985) Nature, 316, 452–457.
22) Rombouts, Y., Ewing, E., van de Stadt, L.A., Selman, M.H., Trouw, L.A., Deelder, A.M., Huizinga, T.W., Wuhrer, M., van Schaardenburg, D., Toes, R.E., & Scherer, H.U. (2015) Ann. Rheum. Dis., 74, 234–241.
26) Anthony, R.M., Nimmerjahn, F., Ashline, D.J., Reinhold, V.N., Paulson, J.C., & Ravetch, J.V. (2008) Science, 320, 373–376.
27) Washburn, N., Schwab, I., Ortiz, D., Bhatnagar, N., Lansing, J.C., Medeiros, A., Tyler, S., Mekala, D., Cochran, E., Sarvaiya, H., Garofalo, K., Meccariello, R., Meador, J.W. III, Rutitzky, L., Schultes, B.C., Ling, L., Avery, W., Nimmerjahn, F., Manning, A.M., Kaundinya, G.V., & Bosques, C.J. (2015) Proc. Natl. Acad. Sci. USA, 112, E4339.
28) Anthony, R.M., Wermeling, F., Karlsson, M.C., & Ravetch, J.V. (2008) Proc. Natl. Acad. Sci. USA, 105, 19571–19578.