1)概要
多発性硬化症(multiple sclerosis:MS)は,中枢神経の炎症性脱髄疾患であり,時間的・空間的に病変が多発するのが特徴である.症状は病変部位によって異なり,視力障害,複視,小脳失調,四肢の筋力低下,感覚障害,膀胱直腸障害などである.病初期は再発寛解を繰り返す病型を示すが,ある時期を過ぎると明らかな再発がないにも関わらず病状が進行する二次進行型に移行する.また,再発寛解病型を経ずに進行型病型を呈する一次進行型も知られている1).再発寛解期は病原性T細胞やB細胞が関与する獲得免疫系の急性炎症が主な病態であるのに対し,進行期は自然免疫系による慢性炎症や神経変性が主な病態だと推測されている2).経過が長くなると障害が残り,日常生活動作が著しく低下する症例が少なからず存在する.
2)発症機序
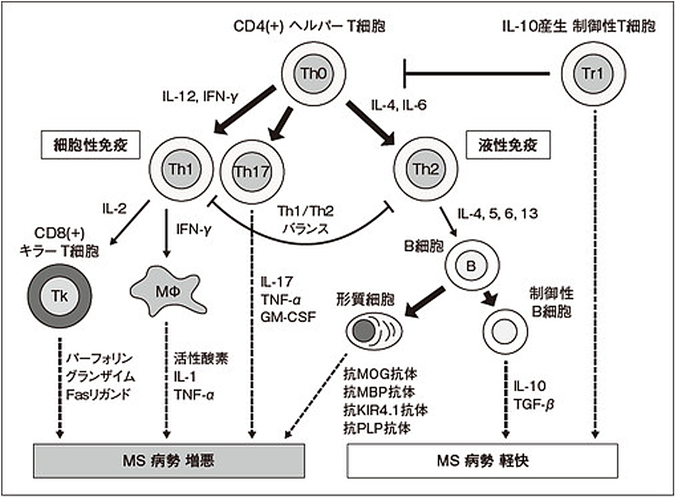
詳細は明らかになっていないが,中枢神経髄鞘タンパク質に対する自己免疫応答に起因し,病巣にリンパ球やマクロファージの浸潤があることや,フィンゴリモドやナタリズマブのような病原性T細胞の中枢神経内への侵入を防ぐ治療薬が有効であることから,病原性T細胞による細胞性免疫が主な病態だと考えられている3).しかし,B細胞を特異的に抑えるリツキシマブ(抗CD20抗体)がMSの疾患活動性を抑制することから,液性免疫もMSの病態に重要な役割を持っていると考えられる(図1).その他のMSの発症機序として,オリゴデンドロサイトの細胞死によって脱髄が生じ,変性した髄鞘を除去するために免疫反応が二次的に生じているという説もある4).MSは,動物モデルとされる実験的自己免疫性脳脊髄炎(experimental autoimmune encephalomyelitis:EAE)の研究によって,これまで詳細に解析されている.
3)疫学
MSの有病率は,有色人種に比べて白人で圧倒的に多いことから遺伝因子が関連しており,日照時間の短さ,活性型ビタミンD3不足,喫煙などの生活習慣が発症リスクになっていることから環境因子の関与も指摘されている5).わが国の患者数は,平成27年の医療受給者証保持者数が19,645人(視神経脊髄炎を含む)であり,年々増加しているが,食事内容の欧米化をはじめとする環境因子の変化が原因の一つだと指摘されている.
4)診断
診断は,現病歴と神経学的診察により時間的・空間的な病変の多発性を証明し,他の疾患を否定することで確定するが(表1),診断が困難な症例も多く,特にMSと同様に視神経と脊髄に病巣を呈する視神経脊髄炎との鑑別診断は重要である6).視神経脊髄炎は,血清中の抗アクアポリン4抗体が関与する液性免疫が主体の疾患であり,MSとは病態が異なるため,治療法も異なる.
表1 多発性硬化症の診断基準(McDonald診断基準2010年改定版より)6)| 臨床像 | 診断に必要な追加事項 |
|---|
| 2回以上の増悪と2個以上の臨床的他覚的病巣(1回の増悪でも病歴で増悪を示唆するものがあればよい) | なし |
| 2回以上の増悪と1個の臨床的他覚的病巣 | MRIによる「空間的多発性」の証明(注1)または,他の病巣に由来する臨床的増悪 |
| 1回の増悪と2個以上の臨床的他覚的病巣 | MRIによる「時間的多発性」の証明(注2)または,2回目の臨床的増悪 |
| 1回の増悪と1個の臨床的他覚的病巣 (clinically isolated syndrome:CIS) | MRIによる「空間的多発性」の証明(注1)または,他の病巣に由来する臨床的増悪およびMRIによる「時間的多発性」の証明(注2)または,2回目の臨床的増悪 |
| MSを示唆する進行性の増悪(一次性進行型) | 1年間の進行性の増悪を認め,以下の三つうちの二つを満たす(注3) |
他疾患の可能性を検討し完全に除外したうえで,中枢神経内に時間的空間的に病変が多発する炎症性脱髄疾患を証明することによって診断する.
注1 空間的多発性:下記のいずれかを満たす.
1. 異なる領域による二つの臨床症状.
2. MRIにおいて,特徴的な領域(脳室周囲,皮質直下,テント下,脊髄)の2領域以上に一つ以上の無症候性のT2病変.
注2 時間的多発性:下記のいずれかを満たす.
1. 1か月以上の間隔をおいた二つの臨床症状.
2. 発症時(初回)のMRIと比較して,再検したMRIで新たなT2病変の確認.
3. 発症時(初回)のMRIで二つ以上のT2病変があり,一つ以上の造影病変と一つ以上の非造影病変.
注3 一次進行型:1年間慢性的に進行する症状を示す症例で,下記三つのうちの二つを満たす.
1. 脳に9個以上のT2病変または脳の4個以上のT2病変とVEP異常.
2. 二つ以上の脊髄病変.
3. 髄液オリゴクローナルバンド陽性かIgGインデックスの上昇. |
5)治療
MSの治療は急性増悪期治療と再発予防に大きく分けられる.急性期には,ステロイド薬の大量点滴静注(パルス療法)や血漿浄化療法を施行する.再発予防に用いられる疾患修飾薬は,わが国ではインターフェロン(IFN)β注射薬,フィンゴリモド,ナタリズマブ,グラチラマー酢酸塩,フマル酸ジメチルが認可されている.各々に一長一短があるため患者によって使い分ける必要がある7).現時点でMSの再発を完全に制御する治療薬はなく,現在も新規治療薬の開発が続いている.
1)概要
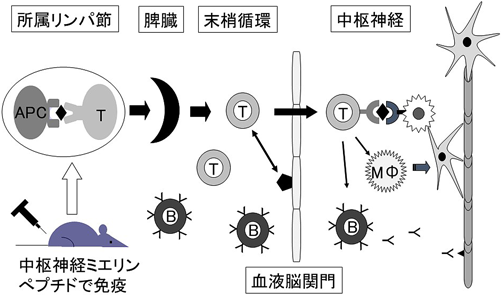
実験的自己免疫性脳脊髄炎(EAE)は,MSの動物モデルとして古くから用いられている.中枢神経髄鞘由来のペプチドをアジュバンドとともに動物に免疫することによって運動麻痺が誘導される(図2).ペプチド特異的T細胞が所属リンパ節や脾臓で増殖し,接着因子やケモカインなどの活性化によって血液脳関門を通過して中枢神経内に侵入する.中枢神経内では,標的抗原の中枢神経を認識し,液性因子の病態も巻き込んで,炎症性脱髄を引き起こす8).
臨床症状のパターンは,動物の種類とペプチドの種類によって異なり,最も用いられるC57/B6マウスにmyelin oligodendrocyte glycoprotein(MOG)(35–55)ペプチドを免疫する方法では,免疫後約1週間で麻痺が始まり,3週間目くらいで麻痺のピークを迎え,その後,徐々に症状は軽快し,およそ5週間目で麻痺は改善し,寛解に至る.SJLマウスにmyelin proteolipid protein(PLP)(135–151)ペプチドを免疫すると,麻痺がいったん軽快した後,再度増悪する臨床パターンを示し,MSの再発寛解型モデルとして用いられている.この他にもさまざまな動物種とペプチドを用いたモデルが確立されており,EAEは実験期間が約1か月で終了する簡便さもあり,MSの病態解析と新規治療薬開発には欠かせない実験ツールとして確立されている.
2)ヘルパーT細胞
EAEは,抗原特異的T細胞を投与しても誘導することができるため(adoptive transfer EAE),病原性T細胞による細胞性免疫が主要な病態であることは確実であり,これに液性免疫も加わった複雑な自己免疫による病態であると推測されている9).
EAEでは,ミエリン構成タンパク質に対するヘルパーT細胞(Th細胞)はIFN-γ産生性のTh1とIL-17を産生するTh17の関与が重要であり,Th1とTh17の双方が病態促進的に働く細胞性免疫の病態であると認識されている.Th1とTh17の関与はMSでも確認されており(図1参照),関節リウマチ,乾癬,潰瘍性大腸炎,クローン病などの自己免疫疾患でも関与が報告されている10).これらの疾患では,Th2や制御性T(Treg)が病態抑制的に働くとされている.
ヒトゲノム解読が完了し,プロテオーム解析によって翻訳後のタンパク質が機能を獲得するのに糖鎖が重要な役割を持つことが明らかになった.糖鎖は,単糖がグリコシド結合によって連結しており,さまざまな長さが存在する.その糖鎖が,細胞膜を貫通するタンパク質に結合したものがプロテオグリカンや糖タンパク質であり,脂質に結合したものが糖脂質である.糖鎖は,細胞の接着,移動,発生,分化,増殖,アポトーシス,感染,免疫などの生命現象が円滑に営まれるように働いており,糖鎖の異常が生じるとがんや糖尿病のような疾患を引き起こす.また糖鎖は,脳,神経,筋肉などの疾患にも関連していることが知られている11).
糖転移酵素は,糖鎖を合成する酵素であり,個体の発生時期や組織によって異なる糖転移酵素が発現している.この酵素は,糖供与体と呼ばれる糖ヌクレオチドからその糖部分を基質となる受容体に転移する反応を触媒しており,糖鎖を伸長する働きをする12).
糖脂質は,脳神経系の発生や分化に関わり,糖脂質の合成が不完全でも脳組織が形成されるが,成長するに従ってさまざまな疾患が生じる13).糖脂質の合成酵素の異常が精神遅滞,発育不全,けいれん等の重篤な神経疾患を引き起こすことも知られている14).これまでの知見から,再生能力を持たない神経細胞は糖鎖によって保護され,安定性が維持されていると考えられている.糖鎖合成機構研究の進展に伴って,糖脂質合成異常,認知症における糖鎖異常,新規O-Man型糖鎖の発見など,糖鎖異常と神経疾患の関連が明らかにされ,その成果をもとにした治療も試みられている15).
末梢神経免疫疾患と糖脂質との関係については,ギラン・バレー症候群およびその亜型でGM1, GalNAc-GD1a, GD1b, GQ1bなどの複合型糖脂質に対する抗体が血清中で上昇することが知られている.これらの抗体は標的抗原の末梢神経における局在部位に結合して病態に直接関与する16).
一方,中枢神経免疫疾患と糖脂質との関係については,MSのモデル動物であるEAEを用いた検証が行われている.複合型糖脂質のGM2/GD2合成酵素を欠損するマウス(GM2/GD2-KO)は,GM2やGD2の合成が不可能で,より下流に位置するGM1, GD1a, GD1b, GT1bなどの複合型糖脂質も欠損している17).GM2/GD2-KOにEAEを誘導すると,野生型マウス(Wt)と比較して症状の重症度は変わらなかったが,発症が約5日間遅延した(図3A).抗原特異的T細胞を移入して誘導するadoptive transfer EAEでも同様の傾向がみられたが,抗原反応性T細胞の抗原再刺激反応(recall response)は,細胞増殖反応および培養上清のサイトカイン(IL-2, IL-4, IL-5, IL-10, IL-17, IFN-γ, TNF)ともに両群間で差はなかった.また,EAE発症直後の中枢神経内のCD4陽性細胞数を比較したところ,GM2/GD2-KO群ではWt群に比べて有意に細胞数が少なかったことから(図3B),複合型糖脂質の有無はTリンパ球活性化には影響しないが,活性Tリンパ球の血液脳関門透過やミエリンへの接着などに影響を及ぼしている可能性があると考えられた18).
1)プロテオグリカン
プロテオグリカン(PG)は細胞骨格の主要成分であり,保水性の高い長い糖鎖がコアタンパク質に結合した複合体で二糖繰り返し構造となっており,糖鎖の種類によってヒアルロン酸,コンドロイチン硫酸(CS),ヘパラン硫酸,ケラタン硫酸(KS)などに分類される.PGは軟骨や角膜などをはじめ,多くの臓器に存在し,中枢神経系では細胞外マトリックスの主要構成成分として豊富に存在している.中枢神経におけるPGは無秩序なシナプス形成を防ぐための再生阻害因子として機能しており,脳損傷動物モデルを用いた実験ではCSやKSが存在すると神経再生が阻害される19–21).
2)中枢神経免疫疾患への影響
EAEの病態におけるCSおよびKSの関与が報告されている.CSの解析には,CS合成に重要な酵素であるN-acetylglucosaminyltransferase 1(CSGalNAcT1)を欠損したマウス(CS-KO)22)と,CSの6位硫酸化に必要なchondroitin 6-O-sulfate transferase 1(C6ST1)を欠損したマウス(C6ST1-KO)23)を用い,KSの解析には脳でのKS生合成に必要なGlcNAc 6-O-sulfotransferase(GlcNAc6ST)遺伝子を欠損したマウス(KS-KO)24)を用いた.
CS-KOはWtに比べてEAE症状は明らかに軽症であった(図4A).抗原(MOG35–55)特異的リンパ球のrecall responseをみると,CS-KO由来のリンパ球はWt由来のリンパ球よりも細胞増殖反応が軽度であったことから(図4B),CSはリンパ球の活性化,つまりEAE induction phaseにて促進的に働いている.しかし,CSの6位硫酸化が阻害されたC6ST1-KOではWtに比べてEAE症状は逆に重症化し(図5A),抗原特異的Tリンパ球を移入したadoptive transfer EAEでも同じくWtよりも悪化した.recall responseではWtと差がなく,EAE発症直後の中枢神経内のCD4陽性細胞数もC6ST1-KO群とWt群で差がなかったことから(図5B),6位硫酸化されたCS(C-unit CS)は末梢での抗原特異Tリンパ球活性化には関与せず,中枢神経内において病原性リンパ球が神経細胞を攻撃する際(EAE effecter phase)の神経組織の感受性に抑制的に働いている可能性が示唆された.さらに,C6ST1を強制発現させたマウス(C6ST1-Tg)ではEAE症状はWtに比べて軽症化したことから(図5A),C-unit CSはEAEに抑制的に機能していることがあらためて確認された.脳損傷モデルではCSは創傷治癒を遅延させることから,CSは中枢神経内において細胞や物質が無秩序に拡散するのを抑制しているものと推測される.つまり,創傷治癒の場面では神経細胞が早く伸展することを妨げるが,EAEでは病原性リンパ球が中枢神経内に浸潤し拡散するのを抑制している可能性があり,特に6位硫酸化されたCSがその役割を強く持っている可能性がある25).
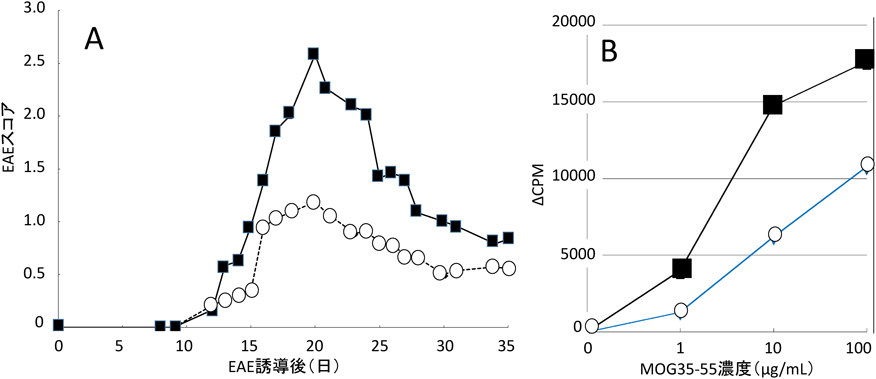
一方,KS-KOではCS-KOと同様にWtと比べてEAE症状は軽症であった(図6A).抗原特異的リンパ球のrecall responseをみると,KS-KO由来のリンパ球はWt由来のリンパ球よりも細胞増殖反応が軽度であり(図6B),KSはCSと同様にEAE induction phaseにおいてリンパ球の活性化に促進的に働いていることが示唆された26).
このように,GAGはCSやKSのようにEAEの病態に促進的に働くものと,C-unit CSのように抑制的に働くものがあることが明らかになった(表2).GAGの合成や生理活性を修飾することでEAEを治療することができる可能性があり,GAGの糖転移酵素の調節はMSをはじめとする神経免疫疾患の新たな治療標的として期待される.
3)腸管微生物を介した役割
近年,GAGはさまざまなサプリメントが市販されており,グルコサミンやヒアルロン酸などは健康食品でおなじみのPGである.それぞれのPGは由来となる生物や臓器によって組成が異なっており,その特徴と効能についての検証がなされている.Sashinamiらは,サケ由来の軟骨PGがEAEの重症度を軽減したと報告している.サケ軟骨PGを経口摂取させると腸管微生物群が変化し,免疫バランスがTh17を抑制し,調節性T細胞を活性化する方向に働くことによってEAEの病態を抑制するという27).
chondroitin sulfate β-1,4-N-acetylgalactosaminyltransferase-1(ChGn-1)はCS鎖の繰り返し領域を合成する酵素であり,CS鎖合成の開始と伸長の両方を担う活性を持つ.ChGn-1が欠損したマウスでは,脊髄外傷からの創傷治癒が良好であったことから,ChGn-1遺伝子の発現量と神経疾患の臨床過程との関連が検討されている28).末梢神経疾患であるギラン・バレー症候群,慢性炎症性脱髄性多発神経炎,遺伝性運動感覚性ニューロパチー(HMSN)などの患者114名および対照として他の神経疾患の196名のChGn-1遺伝子を調べたところ,ベル麻痺患者においてエクソン5(H234R)の,HMSN患者においてエクソン10(M509R)の新規ミスセンス変異を認めた.これらの変異を導入したChGn-1の可溶性フォームを作製しCOS-1細胞で発現させて変異タンパク質を精製し,抗ChGn-1抗体を用いたウエスタンブロット解析を行ったところ,ChGn-1変異タンパク質の発現が確認されたことから,これらの変異は末梢神経障害の病因と関連する可能性がある29).
中枢神経疾患では,MS患者147名,健常被験者181名に対して,ChGn-1遺伝子の一塩基多型(SNP)を解析したところ,MS患者と対照者の約10%で,コーディングSNP(cSNP:rs140161612)が見いだされた.cSNPは126位のセリンがロイシンへ変化し(p. S126L),このChGn-1変異体タンパク質をCOS-1細胞に発現させると酵素活性は示さなかったが,S126L変異のあるMS患者のうち男性では疾患の進行が遅かったことから,このcSNPはMSの臨床的過程に性差がみられることに関連している可能性が推測された30).
糖転移酵素はMSをはじめとする中枢神経自己免疫疾患の病態に関与しており,糖転移酵素の調節が新たな治療標的となる可能性が示唆された.次の段階として,効率よく安全に糖転移酵素を調節できる中和抗体や阻害薬の開発が必要である.この分野の研究はまだ始まったばかりであり,今後の発展が期待される.
表2 EAEに対するグリコサミノグリカン効果| GAG(糖転移酵素) | KOマウスの症状 | 作用部位 |
|---|
| induction phase | 血液脳関門 | effector phase |
|---|
| CS (GalNAcT1) | 重症化 | ○ | × | ○ |
| C-unit CS (C6ST1) | 軽症化 | × | × | ○ |
| KS (GlcNAc6ST1) | 重症化 | ○ | ○ | ○ |
引用文献References
1) Noseworthy, J., Lucchinetti, C., Rodriguez, M., & Weinshenker, B.G. (2000) N. Engl. J. Med., 343, 938–952.
2) Weiner, H.L. (2009) Ann. Neurol., 65, 239–248.
3) 赤石哲也,中島一郎(2015)医学のあゆみ,255, 363–366.
4) Nakahara, J., Aiso, S., & Suzuki, N. (2009) Expert Opin. Ther. Targets, 13, 1375–1386.
5) 山村隆(2013)医学のあゆみ,246, 1103–1106.
6) Polman, C.H., Reingold, S.C., Banwell, B., Clanet, M., Cohen, J.A., Filippi, M., Fujihara, K., Havrdova, E., Hutchinson, M., Kappos, L., Lublin, F.D., Montalban, X., O’Connor, P., Sandberg-Wollheim, M., Thompson, A.J., Waubant, E., Weinshenker, B., & Wolinsky, J.S. (2011) Ann. Neurol., 69, 292–302.
7) 宮本勝一(2013)日本臨床,71, 817–821.
8) 宮本勝一,楠進(2016)医学のあゆみ,259, 154–158.
9) 宮本勝一(2002)炎症と免疫,10, 529–535.
10) 佐藤和貴郎,山村隆(2012)Annual Review 神経2012(鈴木則宏他編),pp. 253–260, 中外医学社.
11) 古川鋼一,近藤裕史,大海雄介,大川祐樹,古川圭子(2013)病理と臨床,31, 826–831.
12) Kitagawa, H. (2014) Biol. Pharm. Bull., 37, 1705–1712.
13) Takamiya, K., Yamamoto, A., Furukawa, K., Yamashiro, S., Shin, M., Okada, M., Fukumoto, S., Haraguchi, M., Takeda, N., Fujimura, K., Sakae, M., Kishikawa, M., Shiku, H., Furukawa, K., & Aizawa, S. (1996) Proc. Natl. Acad. Sci. USA, 93, 10662–10667.
14) Sugiura, Y., Furukawa, K., Tajima, O., Mii, S., Honda, T., & Furukawa, K. (2005) Neuroscience, 135, 1167–1178.
15) 古川鋼一,大海雄介,大川祐樹,古川圭子,田島織絵(2013)Annual Review 神経2013(鈴木則宏他編),pp. 37–45, 中外医学社.
16) Kusunoki, S. (2016) in Neuroimmunological Diseases (Kusunoki, S. ed.), pp. 153–116., Springer Japan, Tokyo.
17) Furukawa, K., Takamiya, K., & Furukawa, K. (2002) Biochim. Biophys. Acta, 1573, 356–362.
18) Miyamoto, K., Takada, K., Furukawa, K., & Kusunoki, S. (2008) Glycobiology, 18, 408–413.
19) Matsui, H., Ohgomori, T., Natori, T., Miyamoto, K., Kusunoki, S., Sakamoto, K., Ishiguro, N., Imagama, S., & Kadomatsu, K. (2013) Cell Death Dis., 4, e946.
20) Haylock-Jacobs, S., Keough, M.B., Lau, L., & Yong, V.W. (2011) Autoimmun. Rev., 10, 766–772.
21) Lau, L.W., Keough, M.B., Haylock-Jacobs, S., Cua, R., Döring, A., Sloka, S., Stirling, D.P., Rivest, S., & Yong, V.W. (2012) Ann. Neurol., 72, 419–432.
22) Sato, T., Kudo, T., Ikehara, Y., Ogawa, H., Hirano, T., Kiyohara, K., Hagiwara, K., Togayachi, A., Ema, M., Takahashi, S., Kimata, K., Watanabe, H., & Narimatsu, H. (2011) J. Biol. Chem., 286, 5803–5812.
23) Uchimura, K., Kadomatsu, K., Nishimura, H., Muramatsu, H., Nakamura, E., Kurosawa, N., Habuchi, O., El-Fasakhany, F.M., Yoshikai, Y., & Muramatsu, T. (2002) J. Biol. Chem., 277, 1443–1450.
24) Uchimura, K., Kadomatsu, K., El-Fasakhany, F.M., Singer, M.S., Izawa, M., Kannagi, R., Takeda, N., Rosen, S.D., & Muramatsu, T. (2004) J. Biol. Chem., 279, 35001–35008.
25) Miyamoto, K., Tanaka, N., Moriguchi, K., Ueno, R., Kadomatsu, K., Kitagawa, H., & Kusunoki, S. (2014) Glycobiology, 24, 469–475.
26) Ueno, R., Miyamoto, K., Tanaka, N., Moriguchi, K., Kadomatsu, K., & Kusunoki, S. (2015) J. Neurosci. Res., 93, 1874–1880.
27) Sashinami, H., Asano, K., Yoshimura, S., Mori, F., Wakabayashi, K., & Nakane, A. (2012) Life Sci., 91, 1263–1269.
28) Takeuchi, K., Yoshioka, N., Higa-Onaga, S., Watanabe, Y., Miyata, S., Wada, Y., Kudo, C., Okada, M., Ohko, K., Oda, K., Sato, T., Yokoyama, M., Matsushita, N., Nakamura, M., Okano, H., Sakimura, K., Kawano, H., Kitagawa, H., & Igarashi, M. (2013) Nat. Commun., 4, 2740.
29) Saigoh, K., Izumikawa, T., Koike, T., Shimizu, J., Kitagawa, H., & Kusunoki, S. (2011) J. Hum. Genet., 56, 143–146.
30) Saigoh, K., Yoshimura, S., Izumikawa, T., Miyata, S., Tabara, Y., Matsushita, T., Miki, T., Miyamoto, K., Hirano, M., Kitagawa, H., Kira, J., & Kusunoki, S. (2016) Neurosci. Res., 108, 55–59.
著者紹介Author Profile
 宮本 勝一(みやもと かついち)
宮本 勝一(みやもと かついち)近畿大学医学部神経内科准教授.医学博士.
略歴1967年大阪府生まれ.92年近畿大学医学部卒業.大阪赤十字病院,京都大学医学部,国立精神・神経センター,ハーバード大学を経て2004年より現職.
研究テーマと抱負神経免疫疾患の病態解析と新規治療薬開発.主に動物モデルを用いた研究を行っている.
趣味マラソン.
 :接着因子.
:接着因子.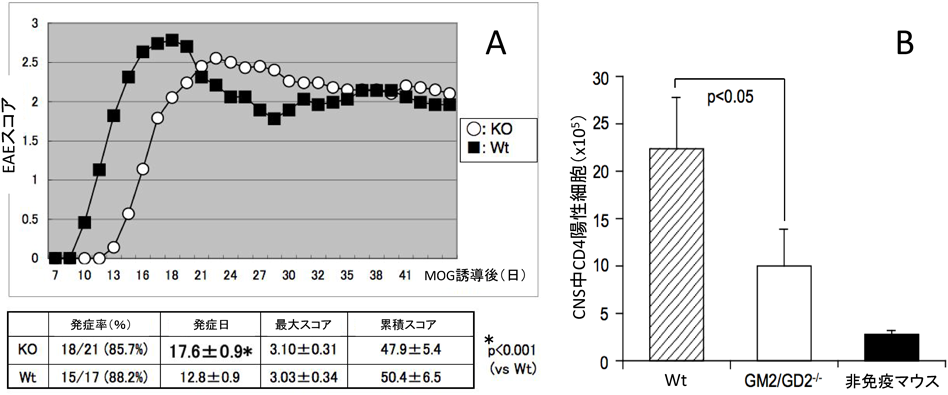
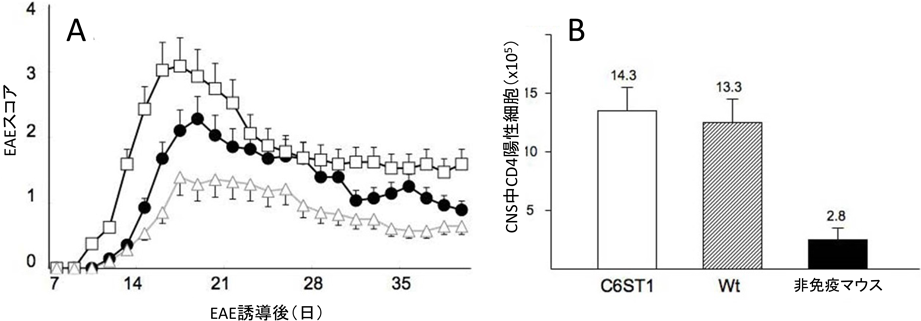
 )で差がなかった(B).
)で差がなかった(B).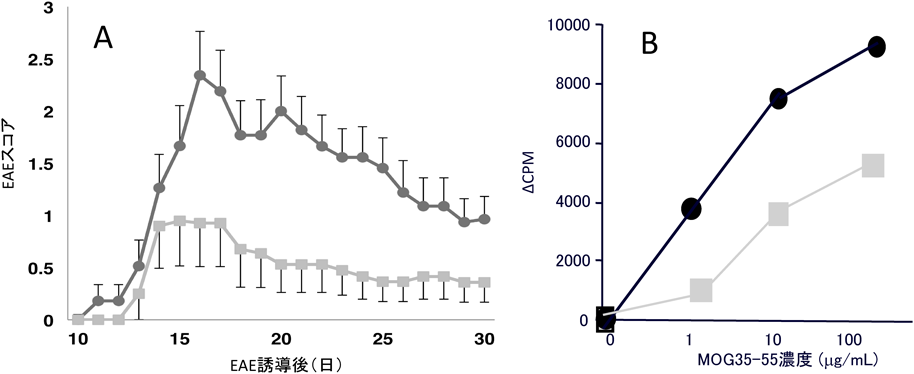
 )にEAEを誘導すると,野生型(Wt:●)に比べて症状は軽症化した(A).抗原(MOG35–55)特異的リンパ球のrecall responseはKS-KO由来のリンパ球(
)にEAEを誘導すると,野生型(Wt:●)に比べて症状は軽症化した(A).抗原(MOG35–55)特異的リンパ球のrecall responseはKS-KO由来のリンパ球(