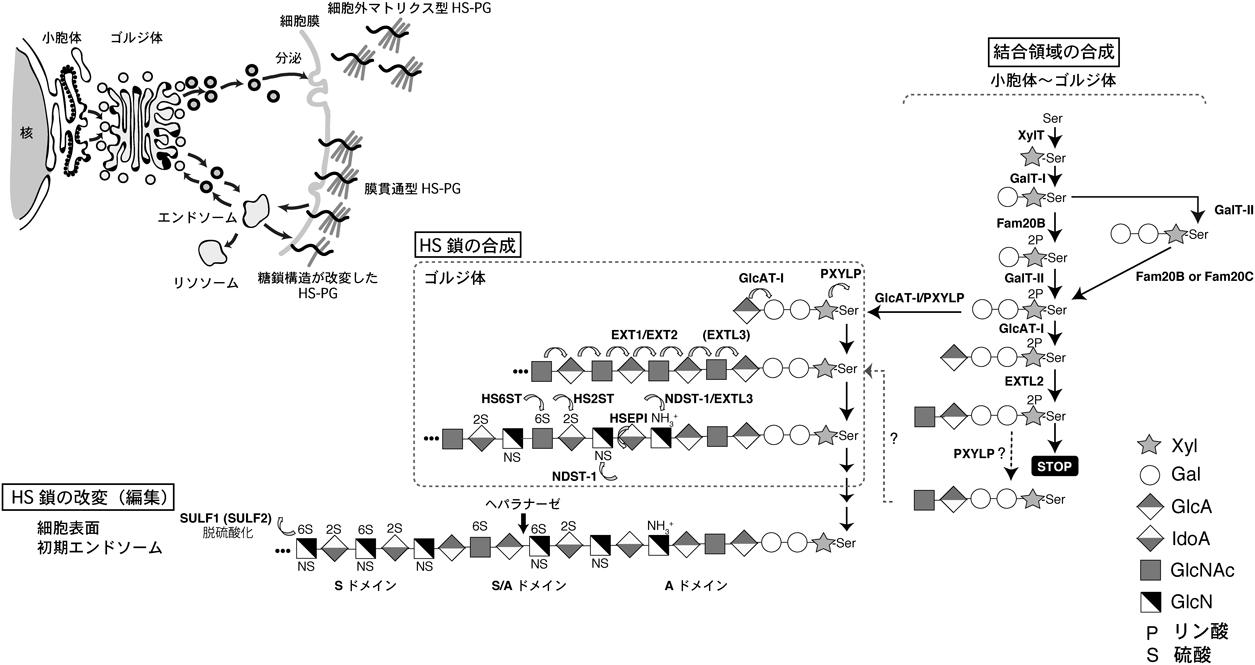
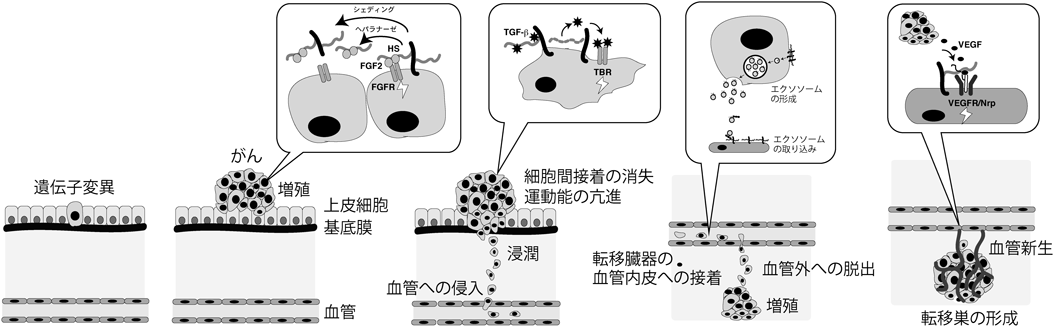
3) Mikami, T. & Kitagawa, H. (2016) Glycoconj. J., doi: 10.1007/s10719-016-9732-9
4) Imamura, M., Higashi, K., Yamaguchi, K., Asakura, K., Furihata, T., Terui, Y., Satake, T., Maegawa, J., Yasumura, K., Ibuki, A., Akase, T., Nishimura, K., Kashiwagi, K., Linhardt, R.J., Igarashi, K., & Toida, T. (2016) Sci. Rep., 6, 33549.
11) Ferreras, C., Rushton, G., Cole, C.L., Babur, M., Telfer, B.A., van Kuppevelt, T.H., Gardiner, J.M., Williams, K.J., Jayson, G.C., & Avizienyte, E. (2012) J. Biol. Chem., 287, 36132–36146.
12) Khurana, A., Beleford, D., He, X., Chien, J., & Shridhar, V. (2013) Am. J. Cancer Res., 3, 34–45.
13) Lai, J., Chien, J., Staub, J., Avula, R., Greene, E.L., Matthews, T.A., Smith, D.I., Kaufmann, S.H., Roberts, L.R., & Shridhar, V. (2003) J. Biol. Chem., 278, 23107–23117.
19) Escobar Galvis, M.L., Jia, J., Zhang, X., Jastrebova, N., Spillmann, D., Gottfridsson, E., van Kuppevelt, T.H., Zcharia, E., Vlodavsky, I., Lindahl, U., & Li, J.P. (2007) Nat. Chem. Biol., 3, 773–778.
21) Yang, Y., Macleod, V., Miao, H.Q., Theus, A., Zhan, F., Shaughnessy, J.D. Jr., Sawyer, J., Li, J.P., Zcharia, E., Vlodavsky, I., & Sanderson, R.D. (2007) J. Biol. Chem., 282, 13326–13333.
26) Li, J., Shworak, N.W., & Simons, M. (2002) J. Cell Sci., 115, 1951–1959.
27) Shintani, Y., Takashima, S., Asano, Y., Kato, H., Liao, Y., Yamazaki, S., Tsukamoto, O., Seguchi, O., Yamamoto, H., Fukushima, T., Sugahara, K., Kitakaze, M., & Hori, M. (2006) EMBO J., 25, 3045–3055.
29) Andres, J.L., DeFalcis, D., Noda, M., & Massague, J. (1992) J. Biol. Chem., 267, 5927–5930.
31) Dhanasekaran, R., Nakamura, I., Hu, C., Chen, G., Oseini, A.M., Seven, E.S., Miamen, A.G., Moser, C.D., Zhou, W., van Kuppevelt, T.H., van Deursen, J.M., Mounajjed, T., Fernandez-Zapico, M.E., & Roberts, L.R. (2015) Hepatology, 61, 1269–1283.
33) Lai, J.P., Sandhu, D.S., Yu, C., Han, T., Moser, C.D., Jackson, K.K., Guerrero, R.B., Aderca, I., Isomoto, H., Garrity-Park, M.M., Zou, H., Shire, A.M., Nagorney, D.M., Sanderson, S.O., Adjei, A.A., Lee, J.S., Thorgeirsson, S.S., & Roberts, L.R. (2008) Hepatology, 47, 1211–1222.
39) Hoshino, A., Costa-Silva, B., Shen, T.L., Rodrigues, G., Hashimoto, A., Mark, M.T., Molina, H., Kohsaka, S., Di Giannatale, A., Ceder, S., Singh, S., Williams, C., Soplop, N., Uryu, K., Pharmer, L., King, T., Bojmar, L., Davies, A.E., Ararso, Y., Zhang, T., Zhang, H., Hernandez, J., Weiss, J.M., Dumont-Cole, V.D., Kramer, K., Wexler, L.H., Narendran, A., Schwartz, G.K., Healey, J.H., Sandstrom, P., Labori, K.J., Kure, E.H., Grandgenett, P.M., Hollingsworth, M.A., de Sousa, M., Kaur, S., Jain, M., Mallya, K., Batra, S.K., Jarnagin, W.R., Brady, M.S., Fodstad, O., Muller, V., Pantel, K., Minn, A.J., Bissell, M.J., Garcia, B.A., Kang, Y., Rajasekhar, V.K., Ghajar, C.M., Matei, I., Peinado, H., Bromberg, J., & Lyden, D. (2015) Nature, 527, 329–335.
40) Baietti, M.F., Zhang, Z., Mortier, E., Melchior, A., Degeest, G., Geeraerts, A., Ivarsson, Y., Depoortere, F., Coomans, C., Vermeiren, E., Zimmermann, P., & David, G. (2012) Nat. Cell Biol., 14, 677–685.
43) Melo, S.A., Luecke, L.B., Kahlert, C., Fernandez, A.F., Gammon, S.T., Kaye, J., LeBleu, V.S., Mittendorf, E.A., Weitz, J., Rahbari, N., Reissfelder, C., Pilarsky, C., Fraga, M.F., Piwnica-Worms, D., & Kalluri, R. (2015) Nature, 523, 177–182.
46) Delgado, M.A., Martinez-Domenech, G., Sarrion, P., Urreizti, R., Zecchini, L., Robledo, H.H., Segura, F., Dodelson de Kremer, R., Balcells, S., Grinberg, D., & Asteggiano, C.G. (2014) Sci. Rep., 4, 6407.