1938年,K. Meyerにより動物起源のアミノ糖を含む複合多糖がムコ多糖と名づけられた.グリコサミノグリカン(glycosaminoglycan:GAG)は,ムコ多糖の系統名として古くから研究されている糖鎖である.GAGは細胞外マトリックスの構成分子として全身に存在する.GAGが保持する組織の支持機能はよく知られている.GAGはその負電荷と生理活性分子との結合から,多くの細胞機能調節にも密接に関わる.
GAGは二糖繰り返し構造を形成する長い直鎖状の多糖である.生体内において,GAGの一種であるヒアルロン酸(hyaluronan:HA)は遊離の糖鎖として存在する.一方,コンドロイチン硫酸(chondroitin sulfate:CS),デルマタン硫酸(dermatan sulfate:DS),ケラタン硫酸(keratan sulfate:KS)およびヘパラン硫酸(heparan sulfate:HS)は,その内部構造に硫酸基修飾がみられ,さまざまな長さの糖側鎖としてコアタンパク質に結合する.GAGに修飾されたタンパク質分子はプロテオグリカン(proteoglycan:PG)と呼ばれる.GAGはPGの生物機能を決定する.
GAGの長さと硫酸化パターンには多様性がみられ,生体内でのGAGの働きも多彩である.また,中枢神経系にも広く分布し神経細胞をはじめ,アストロサイト,ミクログリアなど神経系に存在する細胞の活動にも影響を与える.いずれのGAGも生合成経路はよくわかっており,特定の酵素群が働くことで生合成される.また,GAGを代謝・分解する酵素についても明らかにされている.
本稿では,GAG生合成に関与する酵素遺伝子の欠損あるいは変異が原因になる疾病,およびGAG分解異常が原因になる疾病について概説し,最後に,中枢神経におけるGAGの多様な機能について述べる.
GAGのうちCS,DS,HSの生合成異常が原因となり発症する疾病については,水本らにより詳しく解説されている1).本稿では,CS,DSに加え,KS,HAの生合成異常が起因する疾病について概説する.HSについては,本特集の北川らの稿を参照されたい.
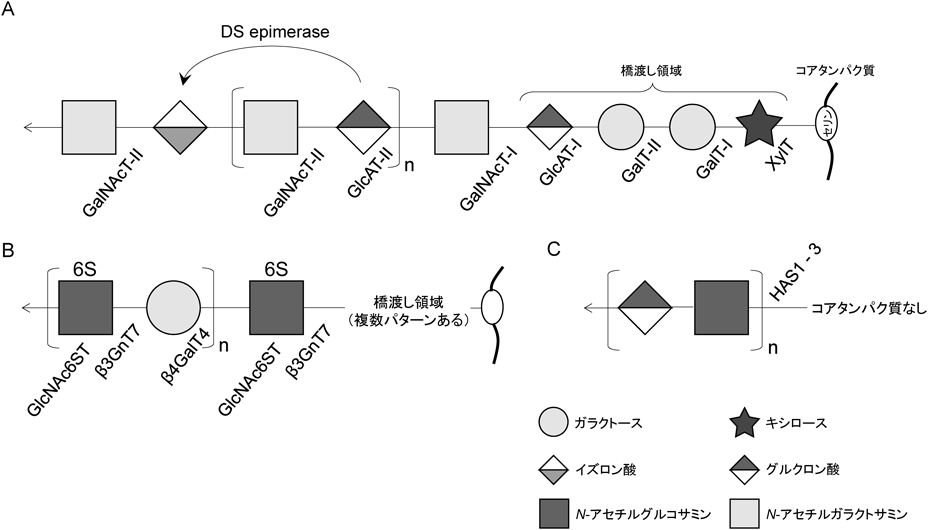
まず,図1に示すようにCS, DSの生合成はコアタンパク質のセリン残基へのキシロース付加から始まる.その後,二つのガラクトースとグルクロン酸が転移されることで,グルクロン酸–ガラクトース–ガラクトース–キシロースという共通四糖橋渡し領域が合成される.そして,繰り返し二糖が転移されていく.CSおよびDSの生合成では,共通四糖のグルクロン酸にN-アセチルガラクトサミン–グルクロン酸が転移されていく.この後,硫酸化反応が起こる.DSの場合,グルクロン酸からイズロン酸へのエピマー化反応が起こる.KS生合成には共通四糖は不要であり,N結合型あるいはO結合型糖鎖の非還元末端に,N-アセチルグルコサミン-ガラクトースが転移されていく.KS鎖の伸長にはN-アセチルグルコサミンのC-6位の硫酸化が必須であると考えられている2).これらのGAG合成酵素異常はさまざまな遺伝性疾患を引き起こす.合成経路を図1,遺伝子の変異あるいは欠損による疾病およびマウスにおける表現型を表1に示した.なお,図1ではCS·HSの硫酸化のステップは省略している.
表1 GAG生合成関連酵素遺伝子と遺伝子変異・欠損による表現型| 変異あるいは欠損酵素 | 遺伝子 | 遺伝子欠損・変異による疾病 | KOマウスの表現型 |
|---|
| キシロース転移酵素(XylT) | XYLT1 | Desbuquois異形成症2型(4) | 低身長,早期骨化(3) |
| β4-ガラクトース転移酵素I (GalT-I) | B4GALT7 | エーラス・ダンロス症候群(早老型1)(5) | — |
| β3-ガラクトース転移酵素II (GalT-II) | B3GALT6 | エーラス・ダンロス症候群(早老型2),脊椎骨端骨幹端異形成症(6) | 胎生致死(−/−),目の形態異常,好酸球の減少(+/−)(33) |
| β3-グルクロン酸転移酵素I (GlcAT-I) | B3GAT3 | ラーセン様症候群(7) | 胎生致死(8) |
| コンドロイチン硫酸N-アセチルガラクトサミン転移酵素(GalNAcT-I) | CSGALNACT1 | 神経障害(9) | 低身長,低体重(10, 11) |
| コンドロイチン合成酵素 | CHSY1 | 軸前性短指症候群(12),神経障害(13) | 軟骨異形成(14) |
| コンドロイチン6-O-硫酸基転移酵素-1 | CHST3 | 軟骨形成異常,脊椎骨端骨幹端異形成症(16),(17) | 脾臓Tリンパ球減少(18) |
| デルマタン4-O-硫酸基転移酵素-1 | CHST14 | エーラス・ダンロス症候群(筋拘縮型)(19) | 低体重,皮膚の脆弱,繁殖能力の低下(21) |
| | 内転母指–内反足症候群(20) | |
| デルマタン硫酸エピメラーゼ1(DS epimerase-1) | DSE | エーラス・ダンロス症候群(筋拘縮型)(22) | 低体重,薄い皮膚(23) |
| デルマタン硫酸エピメラーゼ2(DS epimerase-2) | DSEL | 双極性障害,うつ病性障害(24, 25) | 外見の異常なし(26) |
| β1,3-N-アセチルグルコサミン転移酵素-7(β3GnT7) | B3GNT7 | — | 心肥大,脾臓肥大(−/−♂)(33) |
| 角膜型N-アセチルグルコサミン6-硫酸基転移酵素(GlcNAc6ST-5) | CHST6 | 斑状角膜ジストロフィー(27) | — |
| ヒアルロン酸合成酵素1(Has1) | Has1 | — | 発生,寿命は正常(29) |
| ヒアルロン酸合成酵素2(Has2) | Has2 | — | コンベンショナルKO:胎生致死(31) |
| | | コンディショナルKO(肢芽中胚葉):骨,関節の形成異常(32) |
| ヒアルロン酸合成酵素3(Has3) | Has3 | — | 発生,寿命は正常(30) |
| —:情報,報告なし.文献1)より一部改変して引用.括弧内の数字は,本文引用文献を示す. |
1)キシロース転移酵素I(xylosyltransferase-I:XylT-I)
XYLT1にコードされる糖転移酵素である.このXYLT1欠損マウスでは,軟骨でのGAG量が減少し,低身長になる3).ヒトにおいても同様に,この遺伝子に変異があると,骨格障害がみられる.低身長や関節弛緩などが特徴であるDesbuquois異形成症2型を発症することが報告されており,この疾病発症については,これまでに,XylT-1の5種類の変異が見つかっている4).
2)ガラクトース転移酵素I(galactosyltransferase-I:GalT-I)
この酵素をコードするB4GALT7に変異があると,結合組織に障害が現れるエーラス・ダンロス症候群を発症する.この症候群の早老型1では老いた外見,関節の過伸展,筋弛緩,頭蓋顔面異常,低身長,発育遅延などの症状を引き起こす5).
3)ガラクトース転移酵素II(galactosyltransferase-II:GalT-II)
この酵素をコードするB4GALT6の変異により,エーラス・ダンロス症候群の早老型2や脊椎骨端骨幹端異形成症というような,骨格異常や結合組織障害を発症する.これまでに原因となるGalT-IIの15の遺伝子変異が報告されているが,エーラス・ダンロス症候群と脊椎骨端骨幹端異形成症の発症に共通する変異は見つかっていない6).また,正常なGalT-IIは細胞内において,ゴルジ体に存在する6).一方,変異酵素は核や細胞質に存在することも知られており,細胞内での異常局在が機能不全,疾病発症につながると示唆される.
4)グルクロン酸転移酵素I(glucuronyltransferase-I:GlcAT-I)
この酵素の遺伝子B3GAT3に変異があると,ラーセン様症候群を発症する7).患者は股,膝,肘といった関節を脱臼し多発関節障害を示す.また内反尖足変形や頭蓋顔面異常がみられる.ヒトでもマウスでも,B3GAT3変異はCS, HS量が著しく減少する.マウスでは,この遺伝子のnull欠損は胎生致死となる8).
5)コンドロイチン硫酸N-アセチルガラクトサミン転移酵素(chondroitin sulfate N-acetylgalactosaminyltransferase-I:GalNAcT-I)
この酵素をコードする遺伝子CSGALNACT1にヘテロにミスセンス変異があると,ギラン・バレー症候群などさまざまな神経障害を引き起こす9).この遺伝子のノックアウト(knockout:KO)マウスは,手足,中軸骨格が短くなり,結果として低身長,低体重となる10, 11).
6)コンドロイチン合成酵素(chondroitin synthase)
CHSY1にコードされる酵素で,遺伝子変異がある患者では,軸前性短指症候群がみられる12).また,最近,ヘテロ変異の患者における神経障害が報告された13).KOマウスには,軟骨形成に異常が現れる14).
7)コンドロイチン6-O-硫酸基転移酵素-1(chondroitin 6-O-sulfotransferase-1:C6ST1)
この酵素をコードする遺伝子CHST3における変異は,重篤な軟骨形成異常を示し,脊椎骨端骨幹端異形成症となる15–17).KOマウスでは,脳は正常に発達するが,脾臓におけるTリンパ球の減少が認められる18).
次に,DSについて述べる.DS生合成が正常レベルでないと,エーラス・ダンロス症候群に罹患する.現在までに六つの主病型に分類され,発症頻度はすべてのタイプを合わせて,約5000人に1人と推定されている.
8)デルマタン4-O-硫酸基転移酵素-1(dermatan 4-O-sulfotransferase:D4ST1)
CHST14遺伝子にコードされる,N-アセチルガラクトサミンに硫酸基を付加するこの酵素遺伝子にある変異が起こると,患者は筋拘縮型1のエーラス・ダンロス症候群を発症する.また,内転母指–内反足症候群を発症する遺伝子変異も報告されている19, 20).KOマウスには,低体重,皮膚の脆弱,繁殖能力の低下といった表現型が現れる21).
9)デルマタン硫酸エピメラーゼ1(dermatan sulfate epimerase:DSE)
この酵素は,CSの繰り返し二糖N-アセチルガラクトサミン-グルクロン酸のグルクロン酸残基を異性化し,イズロン酸に変換する.DSE遺伝子にコードされる.この遺伝子に変異があると,エーラス・ダンロス症候群の筋拘縮型2を発症することになる.患者には指,肘,膝など関節に過進展性が現れ,母指,肢の拘縮もみられる.さらには筋疾患も現れる22).KOマウスには,低体重や皮膚が薄くなる表現型が認められる23).
10)デルマタン硫酸エピメラーゼ2(dermatan sulfate epimerase-like:DSEL)
この酵素遺伝子であるDSELに変異があると,双極性障害,うつ病性障害といった精神疾患を発症する24, 25).この遺伝子を欠損したマウスには,外見上の異常はみられない26).
次に,KSおよびHAについて述べる.
11)角膜型N-アセチルグルコサミン6-硫酸基転移酵素(N-acetylglucosamine-6-sulfotransferase 5:GlcNAc6ST-5)
眼の角膜実質が濁り,視力が低下する角膜ジストロフィーの一つに,斑状角膜ジストロフィーがある.斑状角膜ジストロフィーの発症については,この酵素の遺伝子CHST6の変異が原因であることがわかっている27).GlcNAc6STは,先述したKS鎖のN-アセチルグルコサミンのC-6位に硫酸基を付加する酵素である.実際,CHST6に変異がある斑状角膜ジストロフィー患者の角膜実質層の間質には,低硫酸化KSの蓄積が観察される27, 28).その結果,角膜が混濁し透明度が著しく低下し,視力障害を起こす.
12)ヒアルロン酸合成酵素(hyaluronan synthase:Has)
HA生合成異常が起因する疾病の報告はないが,関連酵素遺伝子を欠損したマウスが作製されており,解析されている.HA合成酵素は三つ,Has1, Has2,およびHas3が知られる.Has1およびHas3のKOマウスは,正常に発生し,個体寿命も正常である29, 30).しかし,Has2の全身性KOマウスは胎生致死となり,コンディショナルKOマウスでも,骨や関節の形成異常を示す31, 32).
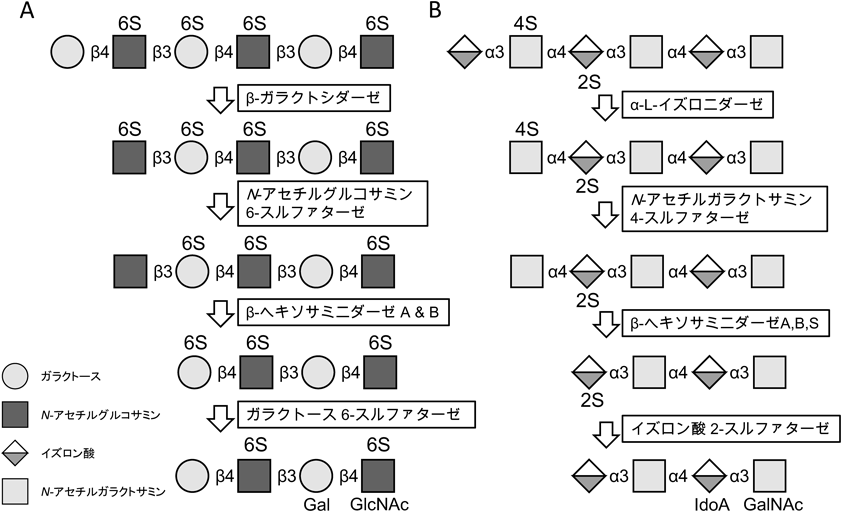
細胞表面および細胞外マトリックスのPGは通常,細胞内へとエンドサイトーシスされ,リソソームへ運ばれて分解される.PG構成成分であるGAGの分解も重要な過程であり,非還元末端よりエキソ型のグリコシダーゼ・スルファターゼにより,段階的に分解される.一例として図2にKSおよびDSの分解経路を示した.KSの分解においては,β-ガラクトシダーゼ34–36),およびβ-ヘキソミニダーゼ37–39)が非還元末端より,それぞれガラクトースおよびN-アセチルグルコサミンを加水分解していく.これらグリコシダーゼは,非還元末端が硫酸化されている場合には作用せず,ガラクトース6-スルファターゼ,N-アセチルグルコサミン6-スルファターゼ40, 41)による脱硫酸化反応を要する(図2A).DS(図2B),CS, HS, HAに関しても同様の分解機構が存在しており,代謝酵素にはある程度の重複性がある.
ムコ多糖症(mucopolysaccharidosis:MPS)は家族性・遺伝性の代謝異常疾患で,これらリソソーム酵素の先天的な欠損・変異により,GAGが細胞内・組織内に異常沈着し,中枢神経障害・骨格異常・関節拘縮・角膜混濁・循環器障害など,種々の病態を引き起こす疾患である.現在では原因酵素・原因遺伝子がほぼすべて明らかになり,11種類の責任酵素が同定され,I型からIX型までの7病型に大分類されている(V型・VIII型は欠番)(表2).ムコ多糖症はわが国においてはリソソーム病として厚生労働省の特定疾患に指定されている.
表2 GAG分解酵素とムコ多糖症| 病型 | 欠損酵素 | 蓄積するグリコサミノグリカン | 中枢神経症状 | 酵素補充療法 | マウスモデル |
|---|
| I | α-L-イズロニダーゼ | デルマタン硫酸 | + | アウドラザイム® | (62) |
| | へパラン硫酸 | | | |
| II | イズロン酸2-スルファターゼ | デルマタン硫酸 | + | エラプレース® | (63) |
| | へパラン硫酸 | | | |
| IIIA | ヘパラン硫酸N-スルファターゼ | へパラン硫酸 | + | | (64) |
| IIIB | α-N-アセチルグルコサミニダーゼ | へパラン硫酸 | + | | (65) |
| IIIC | アセチル-CoA:α-グルコサミニドN-アセチル基転移酵素 | へパラン硫酸 | + | | |
| IIID | N-アセチルグルコサミン6-スルファターゼ | へパラン硫酸 | + | | (66) |
| IVA | N-アセチルガラクトサミン6-スルファターゼ | ケラタン硫酸 | − | ビミジム® | (67) |
| | コンドロイチン硫酸 | | | |
| IVB | β-ガラクトシダーゼ | ケラタン硫酸 | − | | (68) |
| VI | N-アセチルガラクトサミン4-スルファターゼ | デルマタン硫酸 | − | ナグラザイム® | (69) |
| VII | β-グルクロニダーゼ | デルマタン硫酸 | + | | (70) |
| | へパラン硫酸 | | | |
| | コンドロイチン硫酸 | | | |
| IX | ヒアルロニダーゼ | ヒアルロン酸 | + | | (71) |
| 空欄:情報,報告なし.括弧内の数字は,本文引用文献を示す. |
わが国におけるムコ多糖症の発症頻度については,「厚生労働省難治性疾患等政策研究事業に関する調査研究」によると,出生5~6万児に対し1児であるとされる.病型としてはMPS II型が最も多く,全体の半数以上を占める.続いてMPS I型が全体の約20%を占める.MPS IX型はHA分解酵素HYAL1の欠損によって生じる.1996年に初めての症例が報告されて以降42),全世界でも数例の報告にとどまる.以下ではMPS I型,II型,IV型について詳述する.
1)α-L-イズロニダーゼの欠損(MPS I型)
イズロン酸を非還元末端より加水分解する酵素欠損により起こるムコ多糖症である43–45).本酵素はHS・DS双方の代謝に関与するため,患者ではこの二つのGAGが蓄積する.α-L-イズロニダーゼをコードするIDUAは4p16に位置し46),患者では多彩なミスセンス変異やフレームシフトが報告されている47–51).このためか,MPS I型は幅広い臨床像をとり,臨床上の重症度によって,重症型のIH型,軽症型のIS型,これら中間型のIHS型に分類される.重症型のIH型は1歳前後で,骨格系の異常や感染症の反復,臍帯・鼠経ヘルニア,肝脾腫症状を呈する.巨舌も特徴的である.角膜混濁,聴覚障害も出現する.中枢神経症状は通常2歳までに出現する.生命予後はきわめて悪く,心不全,呼吸器感染症,閉塞性肺疾患により,通常小児期に死亡する.軽症型のIS型では関節拘縮,角膜混濁,弁膜症などを呈するものの,中枢神経系症状はきわめて少ない.通常5歳前後で症状が出現し,10歳以降に診断される.生命予後もよく,成人し通常の社会生活を送れるケースも少なくない.
2)イズロン酸2-スルファターゼの欠損(MPS II型)
わが国のムコ多糖症において最も多い病型であり,1973年に明らかにされた52).本酵素はXq28に存在するIDS遺伝子によりコードされる脱硫酸化酵素であるが53–56),患者ではこの遺伝子上に欠失や転座がみられ,酵素活性が大きく減弱する57, 58).ムコ多糖症は常染色体劣性遺伝の形式であるが,MPS II型のみ伴性劣性遺伝である.本酵素はHS·DS中のイズロン酸2-硫酸を基質とするため,患者ではやはり両GAGが異常蓄積し,尿中にも多量に排泄される.蓄積するムコ多糖が同一なためか,I型と非常によく似た臨床像をとる.重症例では,3歳ごろから症状が現れ始める.網膜変性を伴う症例もあるが,角膜混濁は通常みられない.生命予後は悪く,学童期に閉塞性肺疾患,あるいは弁膜症・二次性高血圧による心不全により死亡する.軽症例では,中枢神経症状はなく,成人できるケースも多い.
3)N-アセチルガラクトサミン6-スルファターゼの欠損,β-ガラクトシダーゼの欠損(MPS IV型)
KSの分解異常により引き起こされる疾患で,N-アセチルガラクトサミン6-スルファターゼの欠損によるIVA型と41),β-ガラクトシダーゼの欠損によるIVB型に分類されるが,頻度としてはIVA型の方が圧倒的に多い.N-アセチルガラクトサミン6-スルファターゼはN-アセチルガラクトサミン6-硫酸の脱硫酸化に,β-ガラクトシダーゼは非還元末端のガラクトースの加水分解に必須の酵素である.MPS IV型の患児は正常に誕生するものの,2歳前後より症状を呈する.特に骨格異常が強く出現するのが特徴で,低身長・脊椎後弯および側弯・環椎軸椎間の脱臼など多彩な症状を呈する.角膜混濁が強く出現するが,中枢神経障害はないのが特徴である.これらの特徴は,KSの生理的な発現部位をよく反映しているといえる.
4)治療
ムコ多糖症の治療においては,心不全・呼吸障害など,生命予後の増悪因子のコントロールと,筋骨格異常,感覚器障害,中枢神経症状など患者の生活の質(QOL)を規定する因子を改善することが重要である.
a.酵素補充療法
多くのリソソーム酵素は糖鎖修飾としてマンノース6-リン酸(M6P)を含んだ高マンノース型糖鎖を持っており,これは細胞内輸送系においてM6P受容体を介してリソソームに局在することに役立つ59, 60).M6P受容体は細胞表面にも存在し,細胞から漏洩あるいは分泌されたリソソーム酵素を回収し,リソソームに再分配している61).この細胞機構を利用し,リソソーム酵素を細胞外へ補充し,細胞に取り込ませ,欠損している酵素活性を補填しようという試みが酵素補充療法である.表2にわが国における酵素補充療法の実態をまとめた.わが国においてはI型(アウドラザイム®),II型(エラブレース®),IVA型(ピミジム®),およびVI型(ナグラザイム®)に対する酵素製剤が承認済である.これら酵素製剤は患者において歩行機能や肺機能,尿中GAG排泄量の改善を期待できるが,酵素製剤は血液脊髄関門および血液脳関門を通過できないため,中枢神経症状の改善は難しい.また患者は定期的な通院を要し,補充酵素に対する抗体が形成されてしまうケースも多い.
b.造血幹細胞移植
健常人からの造血幹細胞を患者に移植し,活性を持った酵素を補充しようというものである.移植血球系から分泌・漏洩したリソソーム酵素が,宿主細胞にM6P受容体を介して取り込まれることを期待する.成功すれば永続的な効果が期待できるものの,やはり中枢神経症状への効果は薄い.また移植片対宿主病(GVHD)のような深刻な副作用を起こす危険性もあり,とりわけ,酵素補充療法の利用できる病型では適応に慎重を期すべきである.
5)マウスモデル
表2に示したように,IIIC型を除き,ほぼすべての病型のムコ多糖症のモデルマウスが樹立されている62–71).多くは責任酵素遺伝子のKOマウスとして作出されている.これらモデルマウスはヒトの病態をよく模倣しており,病態の理解や,治療法の開発に有効である.今後は患者由来のiPS細胞の樹立など,より患者病態に即した研究材料の開発が期待される.
最後に,中枢神経系におけるGAGの多彩な働きについて概説する.近年,GAGが成熟脳の高次神経活動にも関与することが明らかにされつつあり,生理条件下や病理条件下においても神経可塑性を制御する証拠が次々と報告されている.
1)神経可塑性
神経細胞の特徴的な機能の一つである記憶学習機能には,シナプス伝達効率の変化が重要であり,興奮性神経細胞において長期増強が起こることで,伝達効率が上がる.この長期増強には,シナプス後膜におけるAMPA受容体の側方移動とクラスタリングが必須である.Frischknechtらは,細胞外のCSおよびHAがこのAMPA受容体の側方移動を抑制すると報告した72, 73).また,OrlandoらはCSが,シナプスで入力を受ける樹状突起におけるスパインの突出や運動を抑えると報告した74).また,CSやKSは,経験依存的神経可塑性も制御することが知られる.一般的に眼優位性可塑性は,臨界期と呼ばれる生後初期のある決められた期間にしか起きないといわれる.これは,GAGが主要な構成分子である神経細胞の近傍に形成されるperineuronal net(PNN)により,神経の可塑性が抑えられることが一因と考えられている.CSを消化する酵素コンドロイチナーゼABCを投与された成獣ラットでは,本来観察されない眼優位性可塑性が確認できる75).また,CS硫酸化酵素の一つであるCS6ST-1トランスジェニックマウスでは,成獣マウスにおいて眼優位性可塑性が観察される76).KS合成酵素であるGlcNAc6ST-1のKOマウスにおいては,臨界期の眼優位性可塑性に異常がみられる77).さらに,CSは扁桃体において,PNNを形成し,恐怖記憶消去を妨げるといったように,経験依存的神経可塑性も制御すると報告されている78).
2)軸索再生
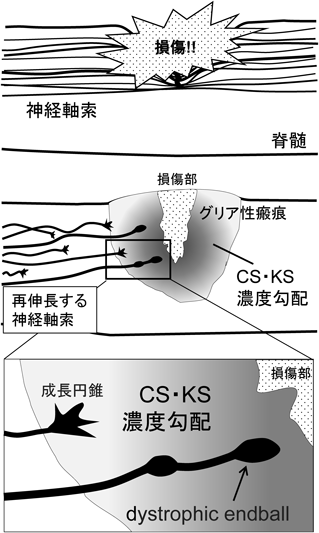
中枢神経系において損傷により異常に生合成され,自発的な神経軸索の再伸長を阻害するGAGについて述べる.脊髄損傷など傷ついた中枢神経系を治療するには,長い神経軸索を再生,再伸長させ,機能する神経回路を再構築させることが唯一の方法であることはいうまでもない79).しかし,損傷部位には強力な軸索再伸長阻害機構が働き,神経損傷は克服できない疾病となっている.これまでに,中枢神経系の軸索再生阻害機構については,生化学的アプローチにより阻害機構に関わるさまざまな分子が同定されてきた.歴史的には,GAGではなくmyelin inhibitorsと呼ばれる分子群が最も注目された時代もあったが,myelin inhibitorsの中枢神経損傷後の軸索再生・機能回復への関与はきわめて限定的という報告がなされた80).これにより,GAGへの注目がなおいっそう高まってきた.脊髄組織において,CSは損傷部位で神経軸索の再伸長を妨げる因子として合成され,軸索再伸長に対して強力なバリアーとなる73, 81, 82).約90年前にCajalは,成熟哺乳類では一度傷ついた中枢神経は,二度と再生しないと記している83).しかし,科学技術の発展とともに,成熟した哺乳類の中枢神経系でも,神経細胞の再生能力はなくなるわけではないことがわかってきた84).これまでに,阻害メカニズムに関する研究調査が広く行われており,重要な報告がなされている.以下にその詳細を述べる.
脊髄組織が損傷すると,グリア性瘢痕ができるというセオリーがSilverの研究グループにより提唱され,現在も広く受入れられている80, 85).このグリア性瘢痕において,活性化アストロサイトにより生成,分泌されたCSPGが軸索の再伸長を妨げる.脊髄損傷したラットに,CSを消化する酵素コンドロイチナーゼABCを投与すると,損傷部を越えて伸びる神経軸索が観察され,運動機能も回復する86).また,CSの受容体として,軸索先端部に発現し機能する受容体型のチロシン脱リン酸化酵素(receptor-type protein tyrosine phosphatase:PTPR)の存在も報告されている.そのサブタイプの1つであるPTPRσ遺伝子のKOマウスは,脊髄損傷後の運動機能回復がよい87).さらに,PTPRσの細胞内にある機能ドメインの脱リン化酵素活性を薬理学的に抑制すると脊髄損傷後の良好な機能回復が観察される88).PTPRσはヘパリン様構造を含むHSとも結合することが明らかにされており,軸索伸長を正に制御する結果も観察されている89).HSがPTPRσに結合すると,PTPRσは多量体を形成し活性を失う.ところが,CSと結合すると,PTPRσは単量体となり,酵素活性を発揮すると考えられている.CSとHSは,軸索伸長に対しては逆の効果を示す80, 89).この現象は非常に興味深く,研究することは学術上の意味も大きい.
CSと同様に,KSもまた損傷後に出現する阻害因子と考えられている90, 91).脊髄損傷,大脳皮質損傷など中枢神経損傷モデルにおいて,ある構造を持ったKSが損傷部位に発現することが報告されている90).また,マウス中枢神経由来のKSPGは,培養された小脳顆粒細胞の神経突起伸長を強力に阻害し,この阻害効果はKS消化酵素ケラタナーゼの処理により消失する91).KS生合成を担うGlcNAc6ST-1の遺伝子KOマウスは,大脳皮質損傷後の損傷部周辺の再伸長する神経突起の数が野生型マウスに比べて増加する92).脊髄を損傷させると,損傷部における軸索再伸長が促進される91).さらに,脊髄損傷ラットを作製し,損傷部にケラタナーゼ処理を施すと,軸索の再伸長が促進され,運動機能への治療効果も確認された93).しかし,軸索再伸長への阻害メカニズムにおけるKSの分子メカニズムについては不明な点が多く,今後の研究発展が期待されている.
CSおよびKSの阻害効果について,細胞内シグナル経路はほとんど明らかにされていない.一方,この二つのGAGを受容した神経軸索は,非常に特徴的な形態変化を起こすことが知られている94).それは,dystrophic endball(DE)と呼ばれる軸索先端部が大きく膨んだ形態である94).図3には,損傷した脊髄に形成されるグリア性瘢痕とCS・KS濃度勾配,また,そこに出現するDEの模式図を示した.我々のグループでは,細胞体から遠く離れた軸索先端部に現れるこのDEこそが損傷神経の陥る病態であると考え,解析を進めている.これまでに,in vitro培養系においてDEを誘導できる実験系が確立されている94).伸長過程にある神経軸索の先端部では,細胞膜動態,細胞質での細胞骨格分子,細胞接着分子やシグナル分子のダイナミックな変動が盛んに起こっている95, 96).そのため,DEにおける生化学的解析は技術的に困難であることもあり,DEに関する情報は少ない.しかし,DE内で起こる細胞内イベントを洞察することで,少しずつDEについて情報が蓄積されている(筆者ら,投稿中).さらなる研究を通じて,DE形成を解除し脊髄損傷など損傷神経に対する治療法確立につながる新たな発見に期待したい.
3)神経変性
中枢神経系における神経変性疾患とGAGの関係について述べる.運動ニューロンが障害を受け,徐々に身体中の筋肉が動かせなくなる萎縮性側索硬化症(ALS)は,嚥下障害および呼吸不全を伴う重篤な疾患である.日本国内だけでも,1万人近くの患者がおり,難病に指定される.ALSモデルマウス系統が多く樹立されており,原因究明・治療法開発のため広く研究されている.これまでに,ある構造を持つKSが,ALSモデルマウス中枢神経系において発現誘導されることがわかっている.そのKSは,活性化型ミクログリア細胞で発現するO-結合型糖鎖であり,シアル酸およびフコース修飾を受けたものであった97).ALSにおけるKS鎖内部の機能ドメインの発現誘導を示唆する結果であり,ALSの発症との因果関係について,より詳細に解析されるであろう.
最近,アルツハイマー病(AD)発症の主な原因とされる,脳内アミロイドβタンパク質(Aβ)の重合・沈着に深く関わるKS鎖が報告された98).それは,シアル酸修飾を受けたKSで,AD患者およびADモデルマウスの脳において発現亢進が認められた98).このKSはALSと同様,ミクログリア細胞表面に発現すること,またGlcNAc6ST-1によりこのKSが合成されることも明らかにされた98).GlcNAc6ST-1酵素タンパク質もAD患者およびADモデルマウスの脳において発現亢進する.GlcNAc6ST-1遺伝子を欠損したADモデルマウスでは,ミクログリア表面のシアル酸修飾KSは消失し,ミクログリアによるAβ貪食が促進され,アミロイド斑沈着が抑えられた98).この特殊なKSあるいはGlcNAc6ST-1を標的とした新規診断法・治療法の開発が期待される.タンパク質凝集を伴う他のアミロイドーシスの病因病態にもGAGが深く関わることが知られており,本研究結果を応用した研究推進も今後の課題である99).
引用文献References
1) Mizumoto, S., Yamada, S., & Sugahara, K. (2014) BioMed Res. Int., 2014, 495764.
2) Kitayama, K., Hayashida, Y., Nishida, K., & Akama, T.O. (2007) J. Biol. Chem., 282, 30085–30096.
3) Mis, E.K., Liem, K.F. Jr., Kong, Y., Schwartz, N.B., Domowicz, M., & Weatherbee, S.D. (2014) Dev. Biol., 385, 67–82.
4) Bui, C., Huber, C., Tuysuz, B., Alanay, Y., Bole-Feysot, C., Leroy, J.G., Mortier, G., Nitschke, P., Munnich, A., & Cormier-Daire, V. (2014) Am. J. Hum. Genet., 94, 405–414.
5) Quentin, E., Gladen, A., Rodén, L., & Kresse, H. (1990) Proc. Natl. Acad. Sci. USA, 87, 1342–1346.
6) Nakajima, M., Mizumoto, S., Miyake, N., Kogawa, R., Iida, A., Ito, H., Kitoh, H., Hirayama, A., Mitsubuchi, H., Miyazaki, O., Kosaki, R., Horikawa, R., Lai, A., Mendoza-Londono, R., Dupuis, L., Chitayat, D., Howard, A., Leal, G.F., Cavalcanti, D., Tsurusaki, Y., Saitsu, H., Watanabe, S., Lausch, E., Unger, S., Bonafé, L., Ohashi, H., Superti-Furga, A., Matsumoto, N., Sugahara, K., Nishimura, G., & Ikegawa, S. (2013) Am. J. Hum. Genet., 92, 927–934.
7) Baasanjav, S., Al-Gazali, L., Hashiguchi, T., Mizumoto, S., Fischer, B., Horn, D., Seelow, D., Ali, B.R., Aziz, S.A., Langer, R., Saleh, A.A., Becker, C., Nürnberg, G., Cantagrel, V., Gleeson, J.G., Gomez, D., Michel, J.B., Stricker, S., Lindner, T.H., Nürnberg, P., Sugahara, K., Mundlos, S., & Hoffmann, K. (2011) Am. J. Hum. Genet., 89, 15–27.
8) Izumikawa, T., Kanagawa, N., Watamoto, Y., Okada, M., Saeki, M., Sakano, M., Sugahara, K., Sugihara, K., Asano, M., & Kitagawa, H. (2010) J. Biol. Chem., 285, 12190–12196.
9) Saigoh, K., Izumikawa, T., Koike, T., Shimizu, J., Kitagawa, H., & Kusunoki, S. (2011) J. Hum. Genet., 56, 143–146.
10) Watanabe, Y., Takeuchi, K., Higa-Onaga, S., Sato, M., Tsujita, M., Abe, M., Natsume, R., Li, M., Furuichi, T., Saeki, M., Izumikawa, T., Hasegawa, A., Yokoyama, M., Ikegawa, S., Sakimura, K., Amizuka, N., Kitagawa, H., & Igarashi, M. (2010) Biochem. J., 432, 47–55.
11) Ema, M., Takahashi, S., Kimata, K., Watanabe, H., & Narimatsu, H. (2011) J. Biol. Chem., 286, 5803–5812.
12) Li, Y., Laue, K., Temtamy, S., Aglan, M., Kotan, L.D., Yigit, G., Canan, H., Pawlik, B., Nürnberg, G., Wakeling, E.L., Quarrell, O.W., Baessmann, I., Lanktree, M.B., Yilmaz, M., Hegele, R.A., Amr, K., May, K.W., Nürnberg, P., Topaloglu, A.K., Hammerschmidt, M., & Wollnik, B. (2010) Am. J. Hum. Genet., 87, 757–767.
13) Izumikawa, T., Saigoh, K., Shimizu, J., Tsuji, S., Kusunoki, S., & Kitagawa, H. (2013) Biochim. Biophys. Acta, 1830, 4806–4812.
14) Wilson, D.G., Phamluong, K., Lin, W.Y., Barck, K., Carano, R.A., Diehl, L., Peterson, A.S., Martin, F., & Solloway, M.J. (2012) Dev. Biol., 363, 413–425.
15) Rajab, A., Kunze, J., & Mundlos, S. (2004) Am. J. Med. Genet., 126A, 413–419.
16) Thiele, H., Sakano, M., Kitagawa, H., Sugahara, K., Rajab, A., Höhne, W., Ritter, H., Leschik, G., Nürnberg, P., & Mundlos, S. (2004) Proc. Natl. Acad. Sci. USA, 101, 10155–10160.
17) van Roij, M.H., Mizumoto, S., Yamada, S., Morgan, T., Tan-Sindhunata, M.B., Meijers-Heijboer, H., Verbeke, J.I., Markie, D., Sugahara, K., & Robertson, S.P. (2008) Am. J. Med. Genet., 146A, 2376–2384.
18) Uchimura, K., Kadomatsu, K., Nishimura, H., Muramatsu, H., Nakamura, E., Kurosawa, N., Habuchi, O., El-Fasakhany, F.M., Yoshikai, Y., & Muramatsu, T. (2002) J. Biol. Chem., 277, 1443–1450.
19) Kosho, T., Takahashi, J., Ohashi, H., Nishimura, G., Kato, H., & Fukushima, Y. (2005) Am. J. Med. Genet., 138A, 282–287.
20) Malfait, F., Syx, D., Vlummens, P., Symoens, S., Nampoothiri, S., Hermanns-Lê, T., Van Laer, L., & De Paepe, A. (2010) Hum. Mutat., 31, 1233–1239.
21) Akyüz, N., Rost, S., Mehanna, A., Bian, S., Loers, G., Oezen, I., Mishra, B., Hoffmann, K., Guseva, D., Laczynska, E., Irintchev, A., Jakovcevski, I., & Schachner, M. (2013) Exp. Neurol., 247, 517–530.
22) Müller, T., Mizumoto, S., Suresh, I., Komatsu, Y., Vodopiutz, J., Dundar, M., Straub, V., Lingenhel, A., Melmer, A., Lechner, S., Zschocke, J., Sugahara, K., & Janecke, A.R. (2013) Hum. Mol. Genet., 22, 3761–3772.
23) Maccarana, M., Kalamajski, S., Kongsgaard, M., Magnusson, S.P., Oldberg, A., & Malmström, A. (2009) Mol. Cell. Biol., 29, 5517–5528.
24) Goossens, D., Van Gestel, S., Claes, S., De Rijk, P., Souery, D., Massat, I., Van den Bossche, D., Backhovens, H., Mendlewicz, J., Van Broeckhoven, C., & Del-Favero, J. (2003) Mol. Psychiatry, 8, 83–89.
25) Shi, J., Potash, J.B., Knowles, J.A., Weissman, M.M., Coryell, W., Scheftner, W.A., Lawson, W.B., DePaulo, J.R. Jr., Gejman, P.V., Sanders, A.R., Johnson, J.K., Adams, P., Chaudhury, S., Jancic, D., Evgrafov, O., Zvinyatskovskiy, A., Ertman, N., Gladis, M., Neimanas, K., Goodell, M., Hale, N., Ney, N., Verma, R., Mirel, D., Holmans, P., & Levinson, D.F. (2011) Mol. Psychiatry, 16, 193–201.
26) Bartolini, B., Thelin, M.A., Rauch, U., Feinstein, R., Oldberg, A., Malmström, A., & Maccarana, M. (2012) Glycobiology, 22, 1007–1016.
27) Akama, T.O., Nishida, K., Nakayama, J., Watanabe, H., Ozaki, K., Nakamura, T., Dota, A., Kawasaki, S., Inoue, Y., Maeda, N., Yamamoto, S., Fujiwara, T., Thonar, E.J., Shimomura, Y., Kinoshita, S., Tanigami, A., & Fukuda, M.N. (2000) Nat. Genet., 26, 237–241.
28) Saito, T., Nishida, K., Nakayama, J., Akama, T.O., Fukuda, M.N., Watanabe, K., Quantock, A.J., Maeda, N., Watanabe, H., & Tano, Y. (2008) Br. J. Ophthalmol., 92, 1434–1436.
29) Kobayashi, N., Miyoshi, S., Mikami, T., Koyama, H., Kitazawa, M., Takeoka, M., Sano, K., Amano, J., Isogai, Z., Niida, S., Oguri, K., Okayama, M., McDonald, J.A., Kimata, K., Taniguchi, S., & Itano, N. (2010) Cancer Res., 70, 7073–7083.
30) Bai, K.J., Spicer, A.P., Mascarenhas, M.M., Yu, L., Ochoa, C.D., Garg, H.G., & Quinn, D.A. (2005) Am. J. Respir. Crit. Care Med., 172, 92–98.
31) Camenisch, T.D., Spicer, A.P., Brehm-Gibson, T., Biesterfeldt, J., Augustine, M.L., Calabro, A. Jr., Kubalak, S., Klewer, S.E., & McDonald, J.A. (2000) J. Clin. Invest., 106, 349–360.
32) Matsumoto, K., Li, Y., Jakuba, C., Sugiyama, Y., Sayo, T., Okuno, M., Dealy, C.N., Toole, B.P., Takeda, J., Yamaguchi, Y., & Kosher, R.A. (2009) Development, 136, 2825–2835.
33) International Mouse Phenotyping Consortium, http://www.mousephenotype.org/
34) Oshima, A., Tsuji, A., Nagao, Y., Sakuraba, H., & Suzuki, Y. (1988) Biochem. Biophys. Res. Commun., 157, 238–244.
35) Yamamoto, Y., Hake, C.A., Martin, B.M., Kretz, K.A., Ahern-Rindell, A.J., Naylor, S.L., Mudd, M., & O’Brien, J.S. (1990) DNA Cell Biol., 9, 119–127.
36) Morreau, H., Galjart, N.J., Gillemans, N., Willemsen, R., van der Horst, G.T., & d’Azzo, A. (1989) J. Biol. Chem., 264, 20655–20663.
37) Applegarth, D.A. & Bozoian, G. (1972) Clin. Chim. Acta, 39, 269–271.
38) Cantz, M. & Kresse, H. (1974) Eur. J. Biochem., 47, 581–590.
39) Sango, K., McDonald, M.P., Crawley, J.N., Mack, M.L., Tifft, C.J., Skop, E., Starr, C.M., Hoffmann, A., Sandhoff, K., Suzuki, K., & Proia, R.L. (1996) Nat. Genet., 14, 348–352.
40) Fuchs, W., Beck, M., & Kresse, H. (1985) Eur. J. Biochem., 151, 551–556.
41) Tomatsu, S., Fukuda, S., Masue, M., Sukegawa, K., Fukao, T., Yamagishi, A., Hori, T., Iwata, H., Ogawa, T., Nakashima, Y., Hanyu, Y., Hashimoto, T., Titani, K., Oyama, R., Suzuki, M., Yagi, K., Hayashi, Y., & Orii, T. (1991) Biochem. Biophys. Res. Commun., 181, 677–683.
42) Natowicz, M.R., Short, M.P., Wang, Y., Dickersin, G.R., Gebhardt, M.C., Rosenthal, D.I., Sims, K.B., & Rosenberg, A.E. (1996) N. Engl. J. Med., 335, 1029–1033.
43) Barton, R.W. & Neufeld, E.F. (1971) J. Biol. Chem., 246, 7773–7779.
44) Clements, P.R., Brooks, D.A., Saccone, G.T., & Hopwood, J.J. (1985) Eur. J. Biochem., 152, 21–28.
45) Ohshita, T., Sakuda, H., Nakasone, S., & Iwamasa, T. (1989) Eur. J. Biochem., 179, 201–207.
46) Scott, H.S., Guo, X.H., Hopwood, J.J., & Morris, C.P. (1992) Genomics, 13, 1311–1313.
47) Scott, H.S., Litjens, T., Nelson, P.V., Thompson, P.R., Brooks, D.A., Hopwood, J.J., & Morris, C.P. (1993) Am. J. Hum. Genet., 53, 973–986.
48) Scott, H.S., Nelson, P.V., Litjens, T., Hopwood, J.J., & Morris, C.P. (1993) Hum. Mol. Genet., 2, 1471–1473.
49) Clarke, L.A., Nelson, P.V., Warrington, C.L., Morris, C.P., Hopwood, J.J., & Scott, H.S. (1994) Hum. Mutat., 3, 275–282.
50) Bunge, S., Kleijer, W.J., Steglich, C., Beck, M., Zuther, C., Morris, C.P., Schwinger, E., Hopwood, J.J., Scott, H.S., & Gal, A. (1994) Hum. Mol. Genet., 3, 861–866.
51) Yamagishi, A., Tomatsu, S., Fukuda, S., Uchiyama, A., Shimozawa, N., Suzuki, Y., Kondo, N., Sukegawa, K., & Orii, T. (1996) Hum. Mutat., 7, 23–29.
52) Bach, G., Eisenberg, F. Jr., Cantz, M., & Neufeld, E.F. (1973) Proc. Natl. Acad. Sci. USA, 70, 2134–2138.
53) Flomen, R.H., Green, E.P., Green, P.M., Bentley, D.R., & Giannelli, F. (1993) Hum. Mol. Genet., 2, 5–10.
54) Wilson, P.J., Meaney, C.A., Hopwood, J.J., & Morris, C.P. (1993) Genomics, 17, 773–775.
55) Bielicki, J., Freeman, C., Clements, P.R., & Hopwood, J.J. (1990) Biochem. J., 271, 75–86.
56) Froissart, R., Millat, G., Mathieu, M., Bozon, D., & Maire, I. (1995) Biochem. J., 309, 425–430.
57) Wilson, P.J., Morris, C.P., Anson, D.S., Occhiodoro, T., Bielicki, J., Clements, P.R., & Hopwood, J.J. (1990) Proc. Natl. Acad. Sci. USA, 87, 8531–8535.
58) Wehnert, M., Hopwood, J.J., Schröder, W., & Herrmann, F.H. (1992) Hum. Genet., 89, 430–432.
59) Brown, W.J., Goodhouse, J., & Farquhar, M.G. (1986) J. Cell Biol., 103, 1235–1247.
60) Baron, R., Neff, L., Brown, W., Courtoy, P.J., Louvard, D., & Farquhar, M.G. (1988) J. Cell Biol., 106, 1863–1872.
61) Baron, R., Neff, L., Brown, W., Louvard, D., & Courtoy, P.J. (1990) J. Cell Sci., 97, 439–447.
62) Clarke, L.A., Russell, C.S., Pownall, S., Warrington, C.L., Borowski, A., Dimmick, J.E., Toone, J., & Jirik, F.R. (1997) Hum. Mol. Genet., 6, 503–511.
63) Cardone, M., Polito, V.A., Pepe, S., Mann, L., D’Azzo, A., Auricchio, A., Ballabio, A., & Cosma, M.P. (2006) Hum. Mol. Genet., 15, 1225–1236.
64) Bhaumik, M., Muller, V.J., Rozaklis, T., Johnson, L., Dobrenis, K., Bhattacharyya, R., Wurzelmann, S., Finamore, P., Hopwood, J.J., Walkley, S.U., & Stanley, P. (1999) Glycobiology, 9, 1389–1396.
65) Li, H.H., Yu, W.H., Rozengurt, N., Zhao, H.Z., Lyons, K.M., Anagnostaras, S., Fanselow, M.S., Suzuki, K., Vanier, M.T., & Neufeld, E.F. (1999) Proc. Natl. Acad. Sci. USA, 96, 14505–14510.
66) Roca, C., Motas, S., Marcó, S., Ribera, A., Sánchez, V., Sánchez, X., Bertolin, J., León, X., Pérez, J., Garcia, M., Villacampa, P., Ruberte, J., Pujol, A., Haurigot, V., & Bosch, F. (2017) Hum. Mol. Genet., 26, 1535–1551.
67) Tomatsu, S., Orii, K.O., Vogler, C., Nakayama, J., Levy, B., Grubb, J.H., Gutierrez, M.A., Shim, S., Yamaguchi, S., Nishioka, T., Montano, A.M., Noguchi, A., Orii, T., Kondo, N., & Sly, W.S. (2003) Hum. Mol. Genet., 12, 3349–3358.
68) Matsuda, J., Suzuki, O., Oshima, A., Ogura, A., Noguchi, Y., Yamamoto, Y., Asano, T., Takimoto, K., Sukegawa, K., Suzuki, Y., & Naiki, M. (1997) Glycoconj. J., 14, 729–736.
69) Evers, M., Saftig, P., Schmidt, P., Hafner, A., McLoghlin, D.B., Schmahl, W., Hess, B., von Figura, K., & Peters, C. (1996) Proc. Natl. Acad. Sci. USA, 93, 8214–8219.
70) Sands, M.S. & Birkenmeier, E.H. (1993) Proc. Natl. Acad. Sci. USA, 90, 6567–6571.
71) Martin, D.C., Atmuri, V., Hemming, R.J., Farley, J., Mort, J.S., Byers, S., Hombach-Klonisch, S., Csoka, A.B., Stern, R., & Triggs-Raine, B.L. (2008) Hum. Mol. Genet., 17, 1904–1915.
72) Frischknecht, R., Heine, M., Perrais, D., Seidenbecher, C.I., Choquet, D., & Gundelfinger, E.D. (2009) Nat. Neurosci., 12, 897–904.
73) Sakamoto, K. & Kadomatsu, K. (2017) Biochim. Biophys. Acta, in press.
74) Orlando, C., Ster, J., Gerber, U., Fawcett, J.W., & Raineteau, O. (2012) J. Neurosci., 32, 18009–18017, 18017a.
75) Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J.W., & Maffei, L. (2002) Science, 298, 1248–1251.
76) Miyata, S., Komatsu, Y., Yoshimura, Y., Taya, C., & Kitagawa, H. (2012) Nat. Neurosci., 15, 414–422., S1–S2.
77) Takeda-Uchimura, Y., Uchimura, K., Sugimura, T., Yanagawa, Y., Kawasaki, T., Komatsu, Y., & Kadomatsu, K. (2015) Exp. Neurol., 274(Pt B), 145–155.
78) Gogolla, N., Caroni, P., Lüthi, A., & Herry, C. (2009) Science, 325, 1258–1261.
79) Tuszynski, M.H. & Steward, O. (2012) Neuron, 74, 777–791.
80) Lee, J.K., Geoffroy, C.G., Chan, A.F., Tolentino, K.E., Crawford, M.J., Leal, M.A., Kang, B., & Zheng, B. (2010) Neuron, 66, 663–670.
81) McKeon, R.J., Schreiber, R.C., Rudge, J.S., & Silver, J. (1991) J. Neurosci., 11, 3398–3411.
82) Davies, S.J., Goucher, D.R., Doller, C., & Silver, J. (1999) J. Neurosci., 19, 5810–5822.
83) Ramon, Y., Cajal, S., & May, R.M.(1928) in Degeneration and Regeneration of the Nervous System, Oxford University Press, London.
84) Davies, S.J., Fitch, M.T., Memberg, S.P., Hall, A.K., Raisman, G., & Silver, J. (1997) Nature, 390, 680–683.
85) Silver, J. & Miller, J.H. (2004) Nat. Rev. Neurosci., 5, 146–156.
86) Bradbury, E.J., Moon, L.D., Popat, R.J., King, V.R., Bennett, G.S., Patel, P.N., Fawcett, J.W., & McMahon, S.B. (2002) Nature, 416, 636–640.
87) Shen, Y., Tenney, A.P., Busch, S.A., Horn, K.P., Cuascut, F.X., Liu, K., He, Z., Silver, J., & Flanagan, J.G. (2009) Science, 326, 592–596.
88) Lang, B.T., Cregg, J.M., DePaul, M.A., Tran, A.P., Xu, K., Dyck, S.M., Madalena, K.M., Brown, B.P., Weng, Y.L., Li, S., Karimi-Abdolrezaee, S., Busch, S.A., Shen, Y., & Silver, J. (2015) Nature, 518, 404–408.
89) Coles, C.H., Shen, Y., Tenney, A.P., Siebold, C., Sutton, G.C., Lu, W., Gallagher, J.T., Jones, E.Y., Flanagan, J.G., & Aricescu, A.R. (2011) Science, 332, 484–488.
90) Jones, L.L. & Tuszynski, M.H. (2002) J. Neurosci., 22, 4611–4624.
91) Ito, Z., Sakamoto, K., Imagama, S., Matsuyama, Y., Zhang, H., Hirano, K., Ando, K., Yamashita, T., Ishiguro, N., & Kadomatsu, K. (2010) J. Neurosci., 30, 5937–5947.
92) Zhang, H., Muramatsu, T., Murase, A., Yuasa, S., Uchimura, K., & Kadomatsu, K. (2006) Glycobiology, 16, 702–710.
93) Imagama, S., Sakamoto, K., Tauchi, R., Shinjo, R., Ohgomori, T., Ito, Z., Zhang, H., Nishida, Y., Asami, N., Takeshita, S., Sugiura, N., Watanabe, H., Yamashita, T., Ishiguro, N., Matsuyama, Y., & Kadomatsu, K. (2011) J. Neurosci., 31, 17091–17102.
94) Tom, V.J., Steinmetz, M.P., Miller, J.H., Doller, C.M., & Silver, J. (2004) J. Neurosci., 24, 6531–6539.
95) Tojima, T., Hines, J.H., Henley, J.R., & Kamiguchi, H. (2011) Nat. Rev. Neurosci., 12, 191–203.
96) Vitriol, E.A. & Zheng, J.Q. (2012) Neuron, 73, 1068–1081.
97) Foyez, T., Takeda-Uchimura, Y., Ishigaki, S., Narentuya, Zhang, Z., Sobue, G., Kadomatsu, K., & Uchimura, K. (2015) Am. J. Pathol., 185, 3053–3065.
98) Zhang, Z., Takeda-Uchimura, Y., Foyez, T., Ohtake-Niimi, S., Narentuya, Akatsu, H., Nishitsuji, K., Michikawa, M., Wyss-Coray, T., Kadomatsu, K., & Uchimura, K. (2017) Proc. Natl. Acad. Sci. USA, 114, 2947–2954.
99) Nishitsuji, K. & Uchimura, K. (2017) Glycoconj. J., in press.