近年のタンパク質の機能解析は,ノックアウト,ノックダウン,ドミナントネガティブの発現などの手法により行われることが主流である.しかし,どのような状況でタンパク質が活性化され,またどのようになると不活性化されるかはタンパク質の機能を知る上できわめて重要な情報である.特に細胞周期Cdkの場合には,活性制御と機能が密接に関わっており,活性制御の研究から解明されてきた細胞周期制御機構も多い.増殖しない神経細胞でCdk5がどのような活性制御を受けているかを,細胞周期Cdkとの相違に注目しながら,述べることとする.
1)Cdk5の活性化サブユニットp35とp39,サイクリンI
Cdk5はCdk1やCdk2など主要なCdkとのアミノ酸レベルの相同性が約60%である.他のCdkと同様にCdk5も単独ではキナーゼ活性を示さず,活性化には活性化サブユニットを必要とする.細胞周期Cdkが特定のサイクリンで活性化されるように,Cdk5も独自の活性化サブユニットを持つ.それらはp35, p39,サイクリンI(CCNI)である.Cdk5はサイクリンD1, D2とは結合するが活性化されない8).Cdk5を精製した初期の論文の一つで,Munc18が活性化サブユニットである可能性が述べられたが9),その後Munc18はp35に結合するCdk5の基質の一つであることが明らかにされている.
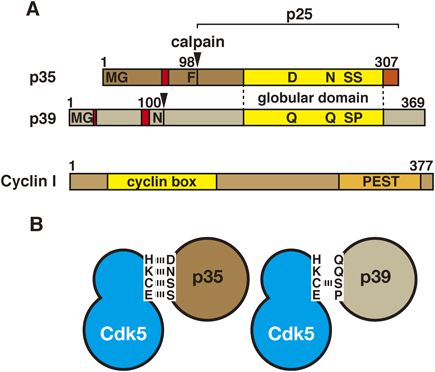
p35は307アミノ酸,p39は369アミノ酸からなり,両者はC末端側のCdk5結合領域(globular domain:GD)のホモロジーが72%とよく似ている(図1).p35の結合はCdk5のactivation loop(T-loop,プロテインキナーゼに共通する領域)の構造を変化させて活性化する.p39にも同様のCdk5活性化機構が想定されるが,Cdk5との結合力にはp35とp39の間で大きな差がある.p35はCdk5と強く結合するのに対して,p39の結合は弱い.p39とCdk5の結合は界面活性剤や高イオン強度で解離する.この結合力の違いはGD内でのCdk5との間で形成する水素結合の数がp39では少なくなっているためと考えられる(図1)10).この弱い結合のため,Cdk5-p39複合体を単離し,その活性を検出することが難しい.後述するようにp35やp39はN末端ミリストイル化を介して膜に結合している.それらを可溶化するためにはTriton X-100やNonidet P-40のような非イオン性界面活性剤を必要とする.しかし,界面活性剤を加えると,Cdk5-p39は解離してしまい,複合体として単離できない.すなわち,Cdk5-p39活性が検出できないことになる.これまでのCdk5研究のほとんどは,Cdk5活性をCdk5-p35に起因するとして得られた結果であるが,その中にはCdk5-p39によるものもあると考えられる.
Cdk5 KOマウスの解析からp35とp39の機能分担の可能性が示唆されている.Cdk5のKOマウスは周産期致死で,脳形成に異常がみられる.p35のKOマウスは大脳皮質の構造に異常がみられるものの,致死ではない.p39のKOマウスには明らかな異常は観察されない.ところが,p35とp39のダブルKOマウスはCdk5 KOマウスと同様に周産期致死となる.このことから,脳におけるCdk5の主要な活性化サブユニットはp35とp39であり,互いに補償できる共通の機能とそれぞれに異なる役割があると考えられている.p35とp39の違いの一部は細胞レベルで示されている.p39 KOマウスから分離した神経細胞では,軸索長の低下,スパインの減少が観察される11).N2a細胞に発現させたCdk5-p35とCdk5-p39の局在は異なっている12).Cdk5-p35が核近傍のエンドソームに局在するのに対して,Cdk5-p39は細胞周囲の葉状仮足に局在する.すなわち,Cdk5-p35は細胞内部で小胞輸送を制御し,Cdk5-p39は細胞膜直下でアクチンフィラメント制御を介して細胞の形態や運動などに関与する機能分担が考えられる.
サイクリンIがCdk5を活性化すると報告されたのは比較的最近である13).サイクリンIはサイクリンに特徴的な配列,サイクリンボックスを有するが,最近まで結合するCdkが不明であった.サイクリンIは腎臓で高い発現を示す.そのKOマウスでは腎臓糸球体の硬化が生後に進行する14).サイクリンIはCdk5の活性化を介し糸球体蛸足細胞の生存に促進的に働いているとのことである.我々もサイクリンIによるCdk5の活性を検証したことがあるが,p35ほど強く活性化する結果は得られなかった15).最近,ヒトでサイクリンIのホモログであるサイクリンI-like(CCNI2)が,CCNIよりも強く,p35と同程度にCdk5を活性化することが報告された16).CCNIが核局在であるのに対して,CCNI2は細胞質の細胞膜近傍に存在することから,両者の機能は異なると考えられる.サイクリンI(またはサイクリンI-like)は普遍的に発現しているので,脳以外の組織での主要なCdk5活性化サブユニットの可能性があるが,その活性化機構や基質などは今後の課題である.
2)Cdk5活性にCdk5の翻訳後修飾は関与しない
細胞周期Cdkでは,サイクリンの結合に加えて,Cdk自体のリン酸化で活性が調節されている.Cdk1はCAK(Cdk-activating kinase)によるT-loop内のThr161のリン酸化を活性化に必要とする.Cdk5にもリン酸化部位(Ser159)は保存されており,CKIIがSer159をリン酸化し活性化するという報告はあるが17),Ser159のリン酸化がなくてもCdk5-p35は十分な活性を示す18).p35の結合がT-loopの位置を変え,ATPが結合できるようにすると結晶構造解析から示されている19).Cdk1はWee1, Myt1によるThr14/Tyr15のリン酸化により活性化が抑制される.Cdk5のTyr15も保存されている.2000年にTyr15がc-Ablによってリン酸化され,Cdk5が活性化されると報告されて以来20),Fyn, Srcなどの非受容体型チロシンキナーゼや,TrkB21),EphA422)などの受容体型チロシンキナーゼでリン酸化され,活性化されるとの報告が続いた.その後,多くの論文がTyr15のリン酸化をそのリン酸化抗体によるウェスタンブロッティングで検出し,活性化された証拠として用いている.しかし,これは間違いであると我々は考えている.Tyr15のリン酸化は,活性化サブユニットと結合していないフリーのCdk5でのみ起こり,p35が結合したCdk5ではみられない.活性化サブユニットの結合により,Tyr15のリン酸化は抑制される.さらに,用いられているリン酸化抗体は,Cdk5を過剰発現させた培養細胞系では確かにTyr15をリン酸化依存的に検出できるが,脳抽出液では非特異的な反応により,Tyr15のリン酸化を見極めることはほぼ不可能である.2014年にこのことを報告して以来15),Tyr15のリン酸化をCdk5の活性化の指標とする報告は激減している(そのため,引用されないが).
3)p35の発現調節,合成と分解
上述したようにCdk5活性は活性化サブユニットの発現量に依存している.増殖細胞におけるサイクリンの場合は,細胞周期に依存して合成・蓄積と分解が繰り返されている.細胞周期のない神経細胞でのp35(やp39)の発現も,Cdk5-p35の神経機能に密接に関与しているはずである.p35の合成は,神経栄養因子(NGF, BDNF, NT3),サイトカイン(TNF-α, TGF-β, IL-6),細胞外基質(ラミニン)などによって増加することが報告されている.NGFやBDNFの下流では,MAPキナーゼ経路を介して,最初期遺伝子(immediate early genes:IEGs)のEgr1によってp35の転写が促進されている.また,発生制御に関連するPOUや,GCボックスに依存した転写制御も観察されている.HDAC阻害剤であるバルプロ酸やトリコスタチンAによってp35の転写が抑制される23).これは,ヒストンアセチル化を介したエピジェネティックな転写調節の存在を示唆している.
Cdk5が安定なタンパク質であるのに対して,p35の半減期は数十分と短い.細胞周期Cdk-サイクリンは機能を終えるとサイクリンの分解によって不活性化する.同様なことが神経細胞で起こっているとすれば,分解を引き起こす刺激は,Cdk5活性と神経活動を結びつける重要な要因となる.しかし,そのような研究は意外と少ない.我々は,グルタミン酸(興奮性神経伝達物質)が,p35の分解を誘導し,Cdk5の一時的な不活性化を引き起こすことを観察している24).ただし,すべての神経伝達物質で分解が誘導されるわけではなく,GABAはp35の量に影響を与えなかった.逆にグルタミン酸処理でCdk5は活性化するという報告もある.グルタミン酸は細胞毒性もあり(後述するp25の生成),グルタミン酸の濃度や処理時間などにより異なる結果が得られていると考えられる.
p35の分解はユビキチン−プロテアソーム経路によると報告された25).p35のリン酸化(Ser8, Ser91, Thr138)が分解へと導くユビキチン化シグナルと考えられる25).神経細胞でみられるSer8, Thr138のリン酸化はCdk5による自己リン酸化で,マウス脳では胎仔期にリン酸化程度が高く,脳の発達に従って低下し,p35は安定化する26).一方,膵臓ベータ細胞での分解シグナルは,AMPキナーゼファミリーの一つであるSIK2によるSer91のリン酸化である.Ser91がリン酸化されたp35は,E3リガーゼPJA2によってユビキチン化され,プロテアソーム分解を受ける27).神経細胞でも,神経活動によって生じたNOによって,p35がS-ニトロシル化されると,PJA2によりユビキチン化され,プロテアソームによって分解される可能性が示されている28).
一方,p35にはユビキチン化されなくてもプロテアソームによって分解される経路もある29).p35のアミノ酸配列中の23個のリシンをすべてアルギニンに置換して,ユビキチン化されない変異p35(23R)を作製したが(N末端はミリストイル化でブロックされている),このp35–23Rもプロテアソームによって分解された29).ユビキチン化依存・非依存的なプロテアソームによる分解の使い分けは今後の課題である.p39もプロテアソームによって分解されるが,p35よりも安定である30).
4)Cdk5-p35とCdk5-p39は膜結合型キナーゼである
細胞周期Cdkと異なるCdk5の大きな特徴の一つは,その細胞内局在にある.分裂細胞では,DNA複製など細胞周期進行に関わる反応は主に核内で起こる.すなわち,細胞周期を制御するCdkも核内に存在してその役割を果たす.分裂を行わない神経細胞では,DNA複製など細胞周期進行は,逆にその細胞の死を意味する.そのためか,活性のあるCdk5は細胞質にアンカーされ,核には入らないようになっている.Cdk5の細胞質局在は活性化サブユニットp35, p39のN末端ミリストイル化に依存する.p35, p39のN末端配列,MGTVLS,はミリストイル化のコンセンサス配列[MGXXX(S/T)]と一致し,N末端2番目のグリシン(G2)がミリストイル化される31).ミリストイル化は翻訳時に起きる脂質修飾であり,脱ミリストイル化酵素が知られていないことから,不可逆的と考えられている.ミリストイル化されていないp35, p39が存在するかどうかは不明である.もしあったとしても,C末端側に存在する核外排出シグナル(NES)によって核には局在しにくくなっていると考えられる31).ただし,ミリストイル基は炭素数が14と短いため,単独では膜アンカーとしては不十分で,他の要因を必要とする.p35, p39の場合はN末端側に存在する塩基性のLysクラスターがそれを担っていると考えられる.p35は主に細胞質内のエンドソームに局在し,p39は細胞質膜近傍に局在している.この違いはLysクラスターにおける塩基性の強さに依存すると思われる.このように,ミリストイル化による膜アンカーは細胞質への局在を保障するのみでなく,局在する細胞内区画にも関与している.
Cdk5は基質タンパク質の性質をリン酸化により変化させるキナーゼである.すなわち,基質タンパク質の同定がCdk5機能解明の一つのキーである.リン酸化部位のコンセンサス配列はすぐC末端側にプロリンがあるセリンまたはトレオニンで,プロリン指向性のセリン/トレオニンキナーゼの一つである.Cdk5の場合はプロリンのC末端側二つ目のアミノ酸が塩基性(リシン/アルギニン)である配列を強く認識する.Cdk1やCdk2とこの性質は共通している.このため,Cdk5(またはCdk1やCdk2)によってリン酸化されやすい部位を探したり,同定したりするのは比較的容易である.ただし,(S/T)Pのみがリン酸化部位の場合は若干紛らわしい.そのような部位は他のプロリン指向性キナーゼであるMAPキナーゼ,GSK3やDyrkなどでもリン酸化されるからである.そのため,Cdk5の基質と報告されたものが,実際には他のキナーゼによるものであったり,また,複数のキナーゼによってリン酸化されていたりすることもある.
神経活動の本質は細胞膜を介した脱分極による活動電位の伝導と,シナプスにおける神経細胞間の刺激伝達である.Cdk5は細胞質,しかも,細胞膜や膜小器官に結合しているキナーゼと考えると,膜に関連した神経活動におけるCdk5の役割は理解しやすい.そのような観点から(我々が特に関心を持っている)いくつかの細胞機能および神経機能を選んで紹介する.
1)Cdk5による小胞輸送制御——神経突起伸長と神経伝達物質の放出
神経細胞は軸索,樹状突起という極性の異なる長い突起を持つ.突起伸長時には,突起表面拡大のための細胞膜成分の供給を必要とするし,成熟した神経細胞では,膜成分を神経活動に応じて補充,回収している.これらの膜成分の輸送には主にエンドソームが関わるが,Cdk5-p35はこのエンドソーム輸送を制御する.Cdk5-p35は輸送駆動力となるモータータンパク質側と輸送される膜側の両方に作用する.順行性のモータータンパク質であるKIF13Bは,Cdk5によるリン酸化でチャネル分子TRPV1を含む小胞と結合するようになる32).KIF2Aは微小管の脱重合活性を持つが,Cdk5はKIF2Aをリン酸化して,その活性を抑える33).また,逆行性の軸索輸送のモータータンパク質であるダイニンに結合するNDEL1は,Cdk5によってリン酸化され,輸送を促進する34).
以下では主に膜側について説明する.小胞輸送はRab GTPaseによって制御されている.神経突起伸長はさまざまなRabによって制御されることが示されているが,比較的詳細に調べられているのがRab11である.培養細胞ではRab11はリサイクリングエンドソームに局在し,エンドソームが細胞膜へと戻るリサイクリング経路に関わっている.神経細胞では細胞体から突起へ,そして突起内の小胞輸送を制御している.Cdk5-p35はRab11によるエンドソーム輸送を抑制する(図2).その仕組みは,Rab11の上流,下流で働く少なくとも二つの基質を介してである.p35結合タンパク質として見つかったLemur kinase 1(LMTK1,旧名AATYK)は,脳で高発現するSer/Thrキナーゼである35).LMTK1をノックダウン,ノックアウトするとエンドソーム輸送の亢進により神経軸索の伸長が著しく増大する36).Cdk5はLMTK1をリン酸化して,おそらく活性化し,Rab11の過活性化を抑え,突起が適度な速度で伸長するように調節していると考えられる37).
Rab11のエフェクターで,Rab8のGEFであるGRABもCdk5によるリン酸化で活性が制御される38).Cdk5によってリン酸化されたGRABはエンドソーム上の活性型Rab11に結合して,軸索内を末端に向かって輸送される.このときのリン酸化されたGRABはRab8 GEF活性を持たない.軸索末端で脱リン酸化されると,Rab8を活性化して,Rab11が結合していたエンドソームへとリクルートする.これによってエンドソームは軸索内輸送型から細胞膜と融合できる型へと変換できるようになると思われる.
Cdk5のエンドサイトーシス,エキソサイトーシスにおける役割は,前シナプスでの神経伝達物質の放出・取り込みとの関連で研究されている.神経伝達物質の分泌は,電位依存性カルシウムチャネル(voltage-dependent calcium channel:VDCC)からの細胞外カルシウムの流入によって誘導される.Cdk5はP/Q型39),N型40),L型41)をリン酸化できる.前シナプスではP/Q型39)のVDCCをリン酸化してチャネル活性を抑制している.Cdk5によるSynapsin 142),Tomosyn43)のリン酸化は放出可能なシナプス小胞のプールと貯蔵プールとの割合を変化させて,エキソサイトーシスに対して抑制的に働いている.Cdk5はMunc1844),Septin 545)をリン酸化し,Syntaxinとの結合を阻害し,SNARE複合体の形成を抑制し,シナプス小胞のエキソサイトーシスを抑える.エキソサイトーシスによって,前シナプス膜に融合した膜成分は,クラスリン依存性のエンドサイトーシスによって回収される.エンドサイトーシスに関わるAmphiphysin 1, Dynamin 1, Synaptojaninは構成的にCdk5によってリン酸化されており,エンドサイトーシスを抑制している.刺激により脱リン酸化され,阻害が解除されると,エンドサイトーシスにより膜を回収できるようになる46).このようにCdk5は全体として前シナプスでの神経伝達物質の分泌・取り込みをいくつものステップで抑制するように働いている.
軸索ガイダンスでも,伸長方向を決めるエキソサイトーシスとエンドサイトーシスにCdk5が関わっていることが報告されている47).軸索先端が誘因分子に遭遇すると,成長円錐内で局所的に細胞内カルシウム濃度が上がり,Ca2+/CaMKIIが活性化される.Ca2+/CaMKIIの下流でCdk5は活性化され,PIPKIγ90をリン酸化してそのエンドサイトーシス促進活性を阻害する.その結果,誘因物質方向でエキソサイトーシスが優位になり,局所的に膜が供給されるためその方向へ伸長する.
2)Cdk5の後シナプスにおける役割と可塑性
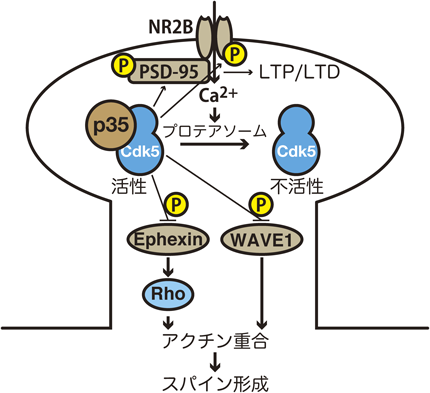
神経伝達物質は後シナプス膜の受容体に結合して興奮を伝達する.グルタミン酸を神経伝達物質とする興奮性の後シナプスはスパインと呼ばれる突起を形成している.スパイン内にはアクチンフィラメントが集積して,スパインの形態を調節している.Cdk5はアクチンフィラメントを介して,スパイン形成を制御している(図3).二つの関与の仕方が示されている.一つはアクチンフィラメントに直接結合するタンパク質をリン酸化する場合,もう一つはRhoファミリーGTPase(RhoA, Rac, Cdc42)を介する場合である.WAVE1はスパインや葉状仮足に存在し,Arp2/3に結合してアクチンフィラメントの重合を促進する因子である.Cdk5によるWAVE1のリン酸化は,WAVE1のアクチンフィラメント重合活性を抑え,結果としてスパインの成熟促進を抑制する48).もう一方のRhoファミリーの制御系では,Racとの関連が強そうである.最初にRacのエフェクターであるPak1にCdk5-p35がRac依存的に結合し,Pak1をリン酸化してそのキナーゼ活性を阻害することが報告された49).その後,Rac1のGEFであるKalirin-7は,Cdk5によるリン酸化でGEF活性が抑制されること,Kalirin-7の過剰発現はスパイン密度を増加させるが,Cdk5でリン酸化できない変異体ではスパインの形態が小さくなることが示された50).一方,RhoAのGEFであるEphexinの場合,Cdk5によるリン酸化でGEF活性が活性化される.RhoAが活性化された結果,スパインは退縮する22).
後シナプスに存在するグルタミン酸受容体にはNMDA型とAMPA型があるが,Cdk5はNMDA型に対して作用するという報告が多い.特にそのサブユニットであるNR2Bの分解を促進したり51),その細胞表面への発現を抑制したりする52).NMDA受容体のエンドサイトーシスには,シナプス後肥厚の構成タンパク質であるPSD-95のクラスタリングが関与しているが,Cdk5はPSD-95をリン酸化し,クラスタリングを制御し,エンドサイトーシスを誘導している53).このようなCdk5による抑制は,NMDA受容体が強く刺激されるとp35の分解を介したCdk5の不活性化により解除される24).すなわち,長期増強(LTP)誘導のような強い刺激では,Cdk5不活性化による抑制解除がポジティブフィードバックとして起こる.
p35KOマウスは脳の層構造に異常はあるが致死ではない.成長するとADHD様の行動を示し54),空間学習にも低下がみられ,長期抑制(LTD)が起きにくくなっていた55).逆にLTPは低頻度の刺激でも起きやすくなっていた24).Cdk5がマウスの行動・記憶に関わっていることが考えられたが,前述したように,Cdk5のコンベンショナルなKOマウスは周産期致死であり,成体脳における役割を知ることはできなかった.コンディショナルなCdk5KOマウスの実現により,記憶・学習実験などが可能となった51).前脳特異的なCdk5KOマウスは,海馬のLTPが促進し,空間学習能力が向上していた51).以上のように,多くの報告はCdk5が神経伝達に抑制的に働くことを示している.これはCdk5が,神経興奮の閾値を決めているという我々の仮説と一致している.
3)タンパク質の発現制御因子としてのCdk5
Cdk5はタンパク質発現を転写,翻訳,タンパク質分解のいずれのレベルにおいても制御している.前述したように,活性型Cdk5は主に細胞質にアンカーされているはずであるが,転写因子や核タンパク質をリン酸化しているという報告も数多くある.具体的にはpRb, p53, MEF2, STAT3, HIF1α, PPARγ, TonEBP/OREBP, CLOCK, Sox6, FOXO1, β-catenin等々である.後述するp35からp25への分解により,膜構造から遊離したCdk5-p25の一部は核内へと移行する.Cdk5が核内で細胞周期調節因子などをリン酸化し,活性化させると,異常な細胞周期進行で神経細胞死が誘導されると考えられる.pRb, p53, MEF2などの細胞周期や細胞の生存などに関わる因子は,核内に移行したCdk5-p25によりリン酸化されるものと考えられる.たとえば線条体ドーパミン細胞を神経毒MPTPで処理すると,Cdk5-p25が生じ,核に移行する.Cdk5-p25は核内でMEF2をリン酸化し,不活性化してMEF2依存的な生存因子の発現を抑えることにより細胞死を誘導する56).一方,CLOCK, Sox6など細胞分化や神経活動に関わる転写因子の場合は,細胞質でリン酸化されるか,または,ミリストイル化されていないp35で活性化されたCdk5によるものであるとすれば説明がつく.たとえば,Cdk5によるCLOCK57),TonEBP/OREBP58)の細胞質でのリン酸化は,それらの核移行を促進する.Sox6は神経分化に関係する転写因子であるが,Cdk5によるリン酸化で分解が誘導され,Cdk5によって発現量が調節されているようである59).神経分化に伴い,活性化サブユニットp35の発現の増加によりCdk5活性は上昇するが,実は神経分化におけるCdk5の役割はあまりよくわかっていない.Cdk5のKOマウス胎仔脳から調製した神経幹細胞では,増殖に異常はみられないが,神経分化が抑制される60).活性化したCdk5-p35が転写因子を介して分化を促進しているとしたら面白いが,まだよくわかっていない.Sox6以外にもCdk5によるリン酸化が,基質タンパク質のユビキチン化のシグナル(デグロン)になることが多く報告されている.p35自身もそうである.Cdk5はホメオスタティックなキナーゼで,その機能の一つはタンパク質の寿命を決めることではないかと考えている.
4)p25によるCdk5の異常活性化とそれにまつわる問題
Cdk5が多くの関心を集めるようになったきっかけの一つは,1999年のアルツハイマー病脳でp35がp25に限定分解され,Cdk5が異常活性化されるという報告である61).この限定分解はカルパインによるものである.カルシウム依存性のプロテアーゼであるカルパインはp35のN末端98アミノ酸を切断し,Cdk5活性化領域からなるp25にする(図1).N末端領域の欠失により,Cdk5-p25は膜構造から遊離し,しかも,安定化(長寿命化)する.このCdk5の異所的かつ持続的な活性がCdk5の異常活性化といわれている.アルツハイマー病脳でのp25レベルの上昇については異論もあり,未決着のままになっている.p25のトランスジェニックマウスも複数のグループで作製されているが,細胞死に関する結論は各グループで異なっている.マウス脳に過剰に発現させると,いったん学習能力などが上昇するが,最終的には神経変性が誘導されるという報告と62),神経変性はみられず,学習行動が改善する63)という報告がある.両者のp25の発現レベルは異なり,発現量によって表現型が異なっている可能性がある.これに関連して,家族性アルツハイマー病でみられる変異APP(アミロイド前駆体タンパク質)を過剰発現するアルツハイマー病のモデルマウスではp25の産生がみられるが64),変異APPをノックインしたマウスではp25の産生がみられないと最近報告され65),Aβ(アミロイドβ)からCdk5の異常活性化という経路については疑問が投げかけられた.
最近,p25に生理的役割があるとする報告があった66).カルパインによる切断部位のアミノ酸を置換すると,p35は限定分解されにくくなる.この変異p35をノックインしたマウスでは,海馬のLTDの誘導や恐怖記憶の消去に不全がみられた.生理的な神経興奮に伴うCa2+濃度の増加が,局所的にカルパインを活性化し,p25を生成し,Cdk5を活性化することはありうると思われる.
神経細胞死・神経変性におけるp25の産生が,原因か結果かはいまだ議論の余地がある.ただし,p25がさまざまなストレスによって生ずるのも確かである.近年では,外傷性脳損傷(TBI)でも,p25の産生によるCdk5の異常活性化が報告されている67).Cdk5-p25が神経細胞死の原因の一つと仮定した場合,Cdk5-p25のみを阻害することは,これら変性や傷害の治療法の一つになる可能性はある.p35/p25のGD(図1)をさらに126アミノ酸まで短くすると,Cdk5に結合するが,活性化できないCIP(Cdk5 inhibitory peptide)と呼ばれる断片となる.さらに,24アミノ酸まで短くしたペプチド(p5)もCdk5を阻害した.p5にTATペプチドを付加したTFP5は,腹腔内に注射しただけで脳内の神経細胞内にも入っていく.前述した前脳特異的にp25を過剰発現するマウスにTFP5を投与したり68),CIPトランスジェニックマウスと交配したりすることで神経変性が抑制された69).p5はin vitroではCdk5-p35もCdk5-p25も阻害するのに対して,in vivoではCdk5-p25を選択的に阻害する.その理由として,in vivoではp35のN末端p10領域に結合したMunc18(前述,当初Cdk5の活性化サブユニットかと誤解されたタンパク質)がTFP5と結合し,p5を吸収してしまい,その阻害活性を抑えるためと説明されている.今後の報告を待ちたいと思う.
4. 非神経系におけるCdk5の働き——最近の話題
Cdk5は神経系でのみ機能すると長らく考えられてきた.しかし,神経細胞ほどではないが,ほとんどの組織・細胞でCdk5は発現している.これまでは,活性化サブユニットが検出されなかったため,機能は疑問視されていたが,最近はCdk5キナーゼ活性も検出され,さまざまな非神経細胞でCdk5が機能していると報告されている.これらの非神経細胞には骨格筋,腎臓,膵臓ベータ細胞,血管内皮細胞,精巣,脂肪細胞,白血球などがある.これらは神経細胞と同様に分化した細胞という特徴を持ち,Cdk5活性と細胞の運動性,形態や生存との関連が示唆されている.前述した腎臓糸球体蛸足細胞のサイクリンIはその一つである.まだ,断片的な報告が多いが,以下では,糖尿病に関わる膵臓ベータ細胞と脂肪細胞,そして,がん細胞におけるCdk5の機能を取り上げる.
1)糖代謝調節——膵臓ベータ細胞と脂肪細胞の場合
Cdk5-p35が膵臓ベータ細胞で発現している.膵臓全体でp35の発現を検出するとわずかであるが,膵島だけにすると十分な発現が検出される.インスリンを分泌する膵島のベータ細胞では,グルコース刺激によってVDCCから細胞内へカルシウムが流入し,インスリン顆粒のエキソサイトーシスが誘導される.Cdk5-p35はL型VDCCをリン酸化してカルシウム流入を抑制し,インスリン分泌を抑えている70).さらに,グルコース刺激はSIK2の分解を抑えてその発現量を高める.SIK2はp35のSer91をリン酸化し,PJA2によるp35のユビキチン化と分解を誘導しCdk5を不活性化する.その結果,VDCCの抑制は解除されインスリン分泌が促進される27).これは神経伝達物質の放出の制御と似たような仕組みである.
脂肪や糖代謝調節において重要な役割をしている脂肪細胞でもCdk5-p35が発現している.インスリンは脂肪細胞でのグルコースの取り込みと糖代謝を促進させるが,インスリン刺激でCdk5が活性化される.その結果,シナプトタグミン様タンパク質E-Syt1がリン酸化され,グルコーストランスポーターであるGLUT4の細胞表面への移行が促進され,脂肪細胞内へグルコースが取り込まれるようになる71).一方,脂肪細胞の成熟を促す核内受容体PPARγは抗糖尿病薬のThiazolidinedioneなどの標的でもある.インスリン感受性を増強するホルモンであるアディポネクチンなどの遺伝子発現はPPARγのSer273のリン酸化によって制御されている.高脂肪食によりp25が生成され,活性化したCdk5-p25が直接Ser273をリン酸化すると一度は報告された72).しかし,Cdk5をノックアウトしたマウスでは逆にSer273のリン酸化が亢進し,インスリン抵抗性を悪化させていた73).これは,Cdk5のノックアウトでERKが活性化され,Ser273をリン酸化している結果だと説明されている.すなわち,Cdk5による脂肪細胞の糖代謝調節はERK-PPARγ経路を介してのようである.
2)がん細胞
p25の過剰発現マウスで脳下垂体の過形成が報告された74).その後,p25の過剰発現マウス(神経変性がみられたマウス)で甲状腺がんが見つかり,p25の過剰発現とがん化との関係が詳細に調べられ75),Cdk5-p25は核内に入り,pRbをリン酸化して甲状腺がんを誘導していることが示された.この報告後,さまざまながん細胞でCdk5の発現・活性の上昇が見つかり,がん化におけるCdk5の役割が注目されるようになっている.pRbのリン酸化は細胞周期のG1/Sチェックポイントの通過に必要であるが,がんの転移・浸潤にもCdk5が関与していると報告されている76).さらに,血管内皮細胞でCdk5がHIF-1αをリン酸化して安定化することが示されているが77),HIF-1αはがん細胞の増殖時の血管新生を誘導する転写因子であり,がん治療のターゲットである.最近,脳腫瘍でCdk5-p35に依存してPD-L1の発現が誘導されているとする報告があった78).PD-L1はT細胞に発現するPD-1に対するリガンドであり,PD-1に結合すると,T細胞の免疫応答が抑制される.がん細胞はPD-L1を発現させて免疫による監視を回避しようとしている.Cdk5はこのPD-1/PD-L1をターゲットとしたがん免疫療法とも関連してくる可能性がある.
引用文献References
1) Ishiguro, K., Takamatsu, M., Tomizawa, K., Omori, A., Takahashi, M., Arioka, M., Uchida, T., & Imahori, K. (1992) J. Biol. Chem., 267, 10897–10901.
2) Lew, J., Beaudette, K., Litwin, C.M., & Wang, J.H. (1992) J. Biol. Chem., 267, 13383–13390.
3) Xiong, Y., Zhang, H., & Beach, D. (1992) Cell, 71, 505–514.
4) Ohshima, T., Ward, J.M., Huh, C.G., Longenecker, G., Veeranna, Pant, H.C., Brady, R.O., Martin, L.J., & Kulkarni, A.B. (1996) Proc. Natl. Acad. Sci. USA, 93, 11173–11178.
5) 大島登志男(2009)蛋白質核酸酵素,54, 796–801.
6) 川内健史(2015)生化学,87, 485–488.
7) 斎藤太郎,久永真市(2013)実験医学,31, 265–270.
8) Guidato, S., McLoughlin, D.M., Grierson, A.J., & Miller, C.C. (1998) J. Neurochem., 70, 335–340.
9) Shetty, K.T., Kaech, S., Link, W.T., Jaffe, H., Flores, C.M., Wray, S., Pant, H.C., & Beushausen, S. (1995) J. Neurochem., 64, 1988–1995.
10) Saito, T., Yano, M., Kawai, Y., Asada, A., Wada, M., Doi, H., & Hisanaga, S. (2013) J. Biol. Chem., 288, 32433–32439.
11) Li, W., Allen, M.E., Rui, Y., Ku, L., Liu, G., Bankston, A.N., Zheng, J.Q., & Feng, Y. (2016) J. Neurosci., 36, 11283–11294.
12) Ito, Y., Asada, A., Kobayashi, H., Takano, T., Sharma, G., Saito, T., Ohta, Y., Amano, M., Kaibuchi, K., & Hisanaga, S. (2014) Mol. Cell. Neurosci., 61, 34–45.
13) Brinkkoetter, P.T., Olivier, P., Wu, J.S., Henderson, S., Krofft, R.D., Pippin, J.W., Hockenbery, D., Roberts, J.M., & Shankland, S.J. (2009) J. Clin. Invest., 119, 3089–3101.
14) Griffin, S.V., Olivier, J.P., Pippin, J.W., Roberts, J.M., & Shankland, S.J. (2006) J. Biol. Chem., 281, 28048–28057.
15) Kobayashi, H., Saito, T., Sato, K., Furusawa, K., Hosokawa, T., Tsutsumi, K., Asada, A., Kamada, S., Ohshima, T., & Hisanaga, S. (2014) J. Biol. Chem., 289, 19627–19636.
16) Liu, C., Zhai, X., Zhao, B., Wang, Y., & Xu, Z. (2017) Sci. Rep., 7, 40979.
17) Sharma, P., Sharma, M., Amin, N.D., Albers, R.W., & Pant, H.C. (1999) Proc. Natl. Acad. Sci. USA, 96, 11156–11160.
18) Poon, R.Y., Lew, J., & Hunter, T. (1997) J. Biol. Chem., 272, 5703–5708.
19) Tarricone, C., Dhavan, R., Peng, J., Areces, L.B., Tsai, L.H., & Musacchio, A. (2001) Mol. Cell, 8, 657–669.
20) Zukerberg, L.R., Patrick, G.N., Nikolic, M., Humbert, S., Wu, C.L., Lanier, L.M., Gertler, F.B., Vidal, M., Van Etten, R.A., & Tsai, L.H. (2000) Neuron, 26, 633–646.
21) Cheung, Z.H., Chin, W.H., Chen, Y., Ng, Y.P., & Ip, N.Y. (2007) PLoS Biol., 5, e63.
22) Fu, W.Y., Chen, Y., Sahin, M., Zhao, X.S., Shi, L., Bikoff, J.B., Lai, K.O., Yung, W.H., Fu, A.K., Greenberg, M.E., & Ip, N.Y. (2007) Nat. Neurosci., 10, 67–76.
23) Takahashi, M., Ishida, M., Saito, T., Ohshima, T., & Hisanaga, S. (2014) Biochem. Biophys. Res. Commun., 447, 678–682.
24) Wei, F.Y., Tomizawa, K., Ohshima, T., Asada, A., Saito, T., Nguyen, C., Bibb, J.A., Ishiguro, K., Kulkarni, A.B., Pant, H.C., Mikoshiba, K., Matsui, H., & Hisanaga, S. (2005) J. Neurochem., 93, 502–512.
25) Patrick, G.N., Zhou, P., Kwon, Y.T., Howley, P.M., & Tsai, L.H. (1998) J. Biol. Chem., 273, 24057–24064.
26) Saito, T., Onuki, R., Fujita, Y., Kusakawa, G., Ishiguro, K., Bibb, J.A., Kishimoto, T., & Hisanaga, S. (2003) J. Neurosci., 23, 1189–1197.
27) Sakamaki, J., Fu, A., Reeks, C., Baird, S., Depatie, C., Al Azzabi, M., Bardeesy, N., Gingras, A.C., Yee, S.P., & Screaton, R.A. (2014) Nat. Cell Biol., 16, 234–244.
28) Zhang, P., Fu, W.Y., Fu, A.K., & Ip, N.Y. (2015) Nat. Commun., 6, 8665.
29) Takasugi, T., Minegishi, S., Asada, A., Saito, T., Kawahara, H., & Hisanaga, S. (2016) J. Biol. Chem., 291, 4649–4657.
30) Minegishi, S., Asada, A., Miyauchi, S., Fuchigami, T., Saito, T., & Hisanaga, S. (2010) Biochemistry, 49, 5482–5493.
31) Asada, A., Yamamoto, N., Gohda, M., Saito, T., Hayashi, N., & Hisanaga, S. (2008) J. Neurochem., 106, 1325–1336.
32) Xing, B.M., Yang, Y.R., Du, J.X., Chen, H.J., Qi, C., Huang, Z.H., Zhang, Y., & Wang, Y. (2012) J. Neurosci., 32, 14709–14721.
33) Ogawa, T. & Hirokawa, N. (2015) Cell Reports, 12, 1774–1788.
34) Sasaki, S., Shionoya, A., Ishida, M., Gambello, M.J., Yingling, J., Wynshaw-Boris, A., & Hirotsune, S. (2000) Neuron, 28, 681–696.
35) Honma, N., Asada, A., Takeshita, S., Enomoto, M., Yamakawa, E., Tsutsumi, K., Saito, T., Satoh, T., Itoh, H., Kaziro, Y., Kishimoto, T., & Hisanaga, S. (2003) Biochem. Biophys. Res. Commun., 310, 398–404.
36) Takano, T., Tomomura, M., Yoshioka, N., Tsutsumi, K., Terasawa, Y., Saito, T., Kawano, H., Kamiguchi, H., Fukuda, M., & Hisanaga, S. (2012) J. Neurosci., 32, 6587–6599.
37) Takano, T., Urushibara, T., Yoshioka, N., Saito, T., Fukuda, M., Tomomura, M., & Hisanaga, S. (2014) Mol. Biol. Cell, 25, 1755–1768.
38) Furusawa, K., Asada, A., Urrutia, P., Gonzalez-Billault, C., Fukuda, M., & Hisanaga, S.I. (2017) J. Neurosci., 37, 790–806.
39) Tomizawa, K., Ohta, J., Matsushita, M., Moriwaki, A., Li, S.T., Takei, K., & Matsui, H. (2002) J. Neurosci., 22, 2590–2597.
40) Su, S.C., Seo, J., Pan, J.Q., Samuels, B.A., Rudenko, A., Ericsson, M., Neve, R.L., Yue, D.T., & Tsai, L.H. (2012) Neuron, 75, 675–687.
41) Furusawa, K., Asada, A., Saito, T., & Hisanaga, S. (2014) J. Neurochem., 130, 498–506.
42) Verstegen, A.M., Tagliatti, E., Lignani, G., Marte, A., Stolero, T., Atias, M., Corradi, A., Valtorta, F., Gitler, D., Onofri, F., Fassio, A., & Benfenati, F. (2014) J. Neurosci., 34, 7266–7280.
43) Cazares, V.A., Njus, M.M., Manly, A., Saldate, J.J., Subramani, A., Ben-Simon, Y., Sutton, M.A., Ashery, U., & Stuenkel, E.L. (2016) J. Neurosci., 36, 11208–11222.
44) Shuang, R., Zhang, L., Fletcher, A., Groblewski, G.E., Pevsner, J., & Stuenkel, E.L. (1998) J. Biol. Chem., 273, 4957–4966.
45) Taniguchi, M., Taoka, M., Itakura, M., Asada, A., Saito, T., Kinoshita, M., Takahashi, M., Isobe, T., & Hisanaga, S. (2007) J. Biol. Chem., 282, 7869–7876.
46) Tomizawa, K., Sunada, S., Lu, Y.F., Oda, Y., Kinuta, M., Ohshima, T., Saito, T., Wei, F.Y., Matsushita, M., Li, S.T., Tsutsui, K., Hisanaga, S., Mikoshiba, K., Takei, K., & Matsui, H. (2003) J. Cell Biol., 163, 813–824.
47) Tojima, T., Itofusa, R., & Kamiguchi, H. (2014) J. Neurosci., 34, 7165–7178.
48) Kim, Y., Sung, J.Y., Ceglia, I., Lee, K.W., Ahn, J.H., Halford, J.M., Kim, A.M., Kwak, S.P., Park, J.B., Ho Ryu, S., Schenck, A., Bardoni, B., Scott, J.D., Nairn, A.C., & Greengard, P. (2006) Nature, 442, 814–817.
49) Nikolic, M., Chou, M.M., Lu, W., Mayer, B.J., & Tsai, L.H. (1998) Nature, 395, 194–198.
50) Ma, X.M., Kiraly, D.D., Gaier, E.D., Wang, Y., Kim, E.J., Levine, E.S., Eipper, B.A., & Mains, R.E. (2008) J. Neurosci., 28, 12368–12382.
51) Hawasli, A.H., Benavides, D.R., Nguyen, C., Kansy, J.W., Hayashi, K., Chambon, P., Greengard, P., Powell, C.M., Cooper, D.C., & Bibb, J.A. (2007) Nat. Neurosci., 10, 880–886.
52) Plattner, F., Hernández, A., Kistler, T.M., Pozo, K., Zhong, P., Yuen, E.Y., Tan, C., Hawasli, A.H., Cooke, S.F., Nishi, A., Guo, A., Wiederhold, T., Yan, Z., & Bibb, J.A. (2014) Neuron, 81, 1070–1083.
53) Morabito, M.A., Sheng, M., & Tsai, L.H. (2004) J. Neurosci., 24, 865–876.
54) Drerup, J.M., Hayashi, K., Cui, H., Mettlach, G.L., Long, M.A., Marvin, M., Sun, X., Goldberg, M.S., Lutter, M., & Bibb, J.A. (2010) Biol. Psychiatry, 68, 1163–1171.
55) Ohshima, T., Ogura, H., Tomizawa, K., Hayashi, K., Suzuki, H., Saito, T., Kamei, H., Nishi, A., Bibb, J.A., Hisanaga, S., Matsui, H., & Mikoshiba, K. (2005) J. Neurochem., 94, 917–925.
56) Smith, P.D., Mount, M.P., Shree, R., Callaghan, S., Slack, R.S., Anisman, H., Vincent, I., Wang, X., Mao, Z., & Park, D.S. (2006) J. Neurosci., 26, 440–447.
57) Kwak, Y., Jeong, J., Lee, S., Park, Y.U., Lee, S.A., Han, D.H., Kim, J.H., Ohshima, T., Mikoshiba, K., Suh, Y.H., Cho, S., & Park, S.K. (2013) J. Biol. Chem., 288, 36878–36889.
58) Gallazzini, M., Heussler, G.E., Kunin, M., Izumi, Y., Burg, M.B., & Ferraris, J.D. (2011) Mol. Biol. Cell, 22, 703–714.
59) Rudrabhatla, P., Utreras, E., Jaffe, H., & Kulkarni, A.B. (2014) PLoS One, 9, e89310.
60) Zheng, Y.L., Li, B.S., Rudrabhatla, P., Shukla, V., Amin, N.D., Maric, D., Kesavapany, S., Kanungo, J., Pareek, T.K., Takahashi, S., Grant, P., Kulkarni, A.B., & Pant, H.C. (2010) Mol. Biol. Cell, 21, 3601–3614.
61) Patrick, G.N., Zukerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., & Tsai, L.H. (1999) Nature, 402, 615–622.
62) Cruz, J.C., Tseng, H.-C., Goldman, J.A., Shih, H., & Tsai, L.H. (2003) Neuron, 40, 471–483.
63) Angelo, M., Plattner, F., Irvine, E.E., & Giese, K.P. (2003) Eur. J. Neurosci., 18, 423–431.
64) Oakley, H., Cole, S.L., Logan, S., Maus, E., Shao, P., Craft, J., Guillozet-Bongaarts, A., Ohno, M., Disterhoft, J., Van Eldik, L., Berry, R., & Vassar, R. (2006) J. Neurosci., 26, 10129–10140.
65) Saito, T., Matsuba, Y., Yamazaki, N., Hashimoto, S., & Saido, T.C. (2016) J. Neurosci., 36, 9933–9936.
66) Seo, J., Giusti-Rodríguez, P., Zhou, Y., Rudenko, A., Cho, S., Ota, K.T., Park, C., Patzke, H., Madabhushi, R., Pan, L., Mungenast, A.E., Guan, J.S., Delalle, I., & Tsai, L.H. (2014) Cell, 157, 486–498.
67) Yousuf, M., Tan, C., Torres-Altoro, M.I., Lu, F.M., Plautz, E., Zhang, S., Takahashi, M., Hernandez, A., Kernie, S.G., Plattner, F., & Bibb, J.A. (2016) J. Neurochem., 138, 317–327.
68) Shukla, V., Seo, J., Binukumar, B.K., Amin, N.D., Reddy, P., Grant, P., Kuntz, S., Kesavapany, S., Steiner, J., Mishra, S.K., Tsai, L.H., & Pant, H.C. (2017) J. Alzheimers Dis., 56, 335–349.
69) Sundaram, J.R., Poore, C.P., Sulaimee, N.H., Pareek, T., Asad, A.B., Rajkumar, R., Cheong, W.F., Wenk, M.R., Dawe, G.S., Chuang, K.H., Pant, H.C., & Kesavapany, S. (2013) J. Neurosci., 33, 334–343.
70) Wei, F.Y., Nagashima, K., Ohshima, T., Saheki, Y., Lu, Y.F., Matsushita, M., Yamada, Y., Mikoshiba, K., Seino, Y., Matsui, H., & Tomizawa, K. (2005) Nat. Med., 11, 1104–1108.
71) Lalioti, V., Muruais, G., Dinarina, A., van Damme, J., Vandekerckhove, J., & Sandoval, I.V. (2009) Proc. Natl. Acad. Sci. USA, 106, 4249–4253.
72) Choi, J.H., Banks, A.S., Estall, J.L., Kajimura, S., Boström, P., Laznik, D., Ruas, J.L., Chalmers, M.J., Kamenecka, T.M., Blüher, M., Griffin, P.R., & Spiegelman, B.M. (2010) Nature, 466, 451–456.
73) Banks, A.S., McAllister, F.E., Camporez, J.P., Zushin, P.J., Jurczak, M.J., Laznik-Bogoslavski, D., Shulman, G.I., Gygi, S.P., & Spiegelman, B.M. (2015) Nature, 517, 391–395.
74) Takashima, A., Murayama, M., Yasutake, K., Takahashi, H., Yokoyama, M., & Ishiguro, K. (2001) Neurosci. Lett., 306, 37–40.
75) Pozo, K., Castro-Rivera, E., Tan, C., Plattner, F., Schwach, G., Siegl, V., Meyer, D., Guo, A., Gundara, J., Mettlach, G., Richer, E., Guevara, J.A., Ning, L., Gupta, A., Hao, G., Tsai, L.H., Sun, X., Antich, P., Sidhu, S., Robinson, B.G., Chen, H., Nwariaku, F.E., Pfragner, R., Richardson, J.A., & Bibb, J.A. (2013) Cancer Cell, 24, 499–511.
76) Strock, C.J., Park, J.I., Nakakura, E.K., Bova, G.S., Isaacs, J.T., Ball, D.W., & Nelkin, B.D. (2006) Cancer Res., 66, 7509–7515.
77) Herzog, J., Ehrlich, S.M., Pfitzer, L., Liebl, J., Fröhlich, T., Arnold, G.J., Mikulits, W., Haider, C., Vollmar, A.M., & Zahler, S. (2016) Oncotarget, 7, 27108–27121.
78) Dorand, R.D., Nthale, J., Myers, J.T., Barkauskas, D.S., Avril, S., Chirieleison, S.M., Pareek, T.K., Abbott, D.W., Stearns, D.S., Letterio, J.J., Huang, A.Y., & Petrosiute, A. (2016) Science, 353, 399–403.
79) Ballif, B.A., Villén, J., Beausoleil, S.A., Schwartz, D., & Gygi, S.P. (2004) Mol. Cell. Proteomics, 3, 1093–1101.