2001年にノーベル医学生理学賞を受賞したLeland Hartwell博士は,1960年代後半に出芽酵母の形態が増殖の進行に従って周期的に変化することに着目し,細胞周期の特定の時期で増殖を停止する多くの変異体,cdc(cell division cycle)変異体を同定した1).cdc7変異体はそれらの一変異体として発見された2).初期の解析より,Cdc7が機能する時点で,複製開始に必要なタンパク質はすべて合成されており,Cdc7は複製開始の直前で機能すると推定された.1980年代にcdc7変異体の責任遺伝子が明らかにされ,セリン・トレオニンキナーゼをコードすることが示され,その機能についての本格的な研究が開始した3).
2. 活性化サブユニットDbf4の発見と,他生物種などにおけるCdc7ホモログの同定
1)Cdc7と相互作用しそのキナーゼ活性に必要とされる活性化因子Dbf4の発見
出芽酵母において,cdc変異体のスクリーニングとは独立に,複製開始過程に異常を示すことが期待されるdbf(dumbbell former,非許容温度で亜鈴型の形態で増殖を停止する)変異体の探索からdbf4変異体が単離されその変異体の責任遺伝子DBF4が同定された.その後,DBF4の過剰発現がcdc7変異を相補すること,さらには生化学的な解析から,Dbf4はCdc7と結合し,そのキナーゼ活性を活性化する活性化サブユニットであることが明らかとなった4–6).
2)他生物種におけるCdc7-Dbf4ホモログ
Cdc7が出芽酵母以外の他の生物種にも存在するかどうかは不明であったが,1995年に筆者らはキナーゼをコードするアミノ酸配列が高く保存されていることに着目し,degenerate PCR(保存されたアミノ酸配列に対する縮重プライマーを用いたPCR)を用いる手法により分裂酵母hsk1+がCdc7のホモログであることを見出し報告した7).この報告を契機にヒトを含む高等生物におけるCdc7タンパク質のホモログが同定され,Cdc7は種を超えて保存されていることが判明した8–10).Dbf4もCdc7と同様に種間で高く保存されており,分裂酵母ではHsk1との相互作用分子の探索,あるいは生化学的探索により,Dbf4の機能的ホモログであるdfp1+/him1+が同定された11–13).また,ヒトにおいても,hsDbf4/ASK(activator of S phase kinase)が発見された14, 15).
3)Cdc7-Dbf4類似タンパク質
分裂酵母にはCdc7と高い相同性を示す類似のキナーゼSpo4(sporulation 4)と,Dbf4と相同性を持つSpo6が存在する.Spo4およびSpo6は減数分裂期のみに発現しており,興味深いことにSpo4はSpo6と複合体を形成する.このSpo4–Spo6キナーゼ複合体はDNA複製には関与せず,第二減数分裂と胞子形成過程に必須の役割を果たす16).いくつかの高等生物においては複数のDbf4ホモログが存在し,たとえば,ヒトとカエルではDrf1/ASKL1が同定されている17–19).カエルの発生初期にはDbf4は発現していないが代わりにDrf1が発現しており,発生が進むにつれてDbf4に置き換わる20).ヒトのDrf1/ASKL1の発現はS期後期からG2期にかけて上昇し,Dbf4と同様にCdc7のキナーゼ活性を上昇させる17, 19).マウスではDrf1/ASKL1分子は同定されておらず,高等動物におけるDrf1/ASKL1の正確な機能は不明である.
3. Cdc7-Dbf4キナーゼ複合体の構造と活性制御
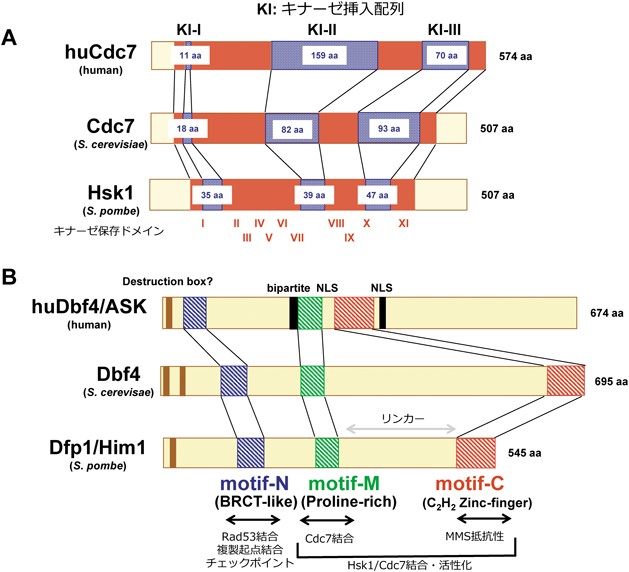
1)Cdc7-Dbf4の保存されたモチーフとそれらの機能(図1)
Cdc7は上述のようにセリン・トレオニン型キナーゼであり,キナーゼ保存配列を有するが,それに加えて保存配列を分断する挿入配列を持つ10, 21, 22)(図1A).長いキナーゼ挿入配列IIおよびIIIは,それぞれ,キナーゼ保存ドメインVIIとVIII, XとXIの間に存在し,種によってはキナーゼ保存ドメインIとIIの間にも短いキナーゼ挿入配列Iが存在する場合がある.キナーゼ挿入配列IIIを欠失すると機能を喪失することから,この配列はCdc7のキナーゼ活性に必要である.また,マウスCdc7にみられる選択的スプライシングアイソフォームはキナーゼ挿入配列II内に32アミノ酸の欠失を持つが,全長とまったく同じ活性を有する10).
一方,活性化サブユニットDbf4には,種間あるいはホモログの構造の比較からDbf4-motif-N, M, Cの三つの保存モチーフが同定されている22, 23)(図1B).Dbf4-motif-MあるいはCのいずれかでも,Cdc7を部分的に活性化するが,完全な活性化には両者が必要である24).この二つのモチーフを連結する領域はリンカーとして機能しており,その長さや配列は活性化に大きな影響を与えない.Dbf4-motif-NはBRCT(BRCA1 C-terminal)様モチーフ1個を有し,その変異は種々のDNA損傷に対する感受性をもたらすが25),Cdc7の活性化には必要ない.増殖にも必須ではないが,Rad53チェックポイントキナーゼとの相互作用に必要とされ,この相互作用の欠損は,複製チェックポイント制御の喪失,あるいはHUやMMSへの感受性をもたらす26, 27).Dbf4-motif-NはRev1のBRCTZモチーフにより機能的に置き換えることが可能である28).Dbf4-motif-Mはプロリンリッチな配列を含み,それのみでCdc7を部分的に活性化することができ,体細胞分裂増殖には十分である29).Dbf4-motif-Cは種間で最も保存された配列で,C2H2タイプのZnフィンガーモチーフを有し,実際にZnを配位したZnフィンガー構造が形成されることが確認されている.減数分裂を介した増殖にはCdc7の完全な活性化が必要であり,Dbf4-motif-Mに加えてDbf4-motif-Cが必要である24).分裂酵母においてDbf4-motif-Cの変異(C末端26アミノ酸が欠失)がrad35変異として同定されている.この変異はアルキル化剤メチルメタンスルホン酸(MMS)に対する感受性をもたらすが,紫外線,電磁放射線,ヒドロキシ尿素(HU)に対する感受性には影響しない29).Dbf4-motif-CのZnフィンガー変異はS期進行の遅延,DNA損傷感受性,紫外線照射の後の変異誘導の低下を示す.またHUやMMSの長期曝露に対する感受性を示した30).高等生物のCdc7の活性化位にはDbf4-motif-MとDbf4-motif-Cが必要で十分であるが,マウスDbf4/ASK欠損細胞を用いた解析から,Dbf4-motif-Nも増殖に必要であることが示された31).動物細胞Dbf4/ASKは,C末端に長いテイル領域を持つが,この領域はCdc7活性化には不要である.ヒトDbf4/ASKのC末端にはセリン・トレオニンを多く含む保存された配列があり,自己リン酸化の標的であるとともに,この配列は分子内相互作用によりCdc7の活性を阻害する32, 33).
2)Cdc7-Dbf4の発現制御
出芽酵母のDbf4および分裂酵母Dfp1/Him1の転写はともに細胞周期進行に伴い周期的に変動する.この転写制御はMluI boxタンパク質Mbp1あるいは転写因子Cdc10に依存する34).またCdc7キナーゼ活性は通常S期に上昇しM期終了時まで活性を保持するがG1期にはその活性が減少する.この細胞周期制御は活性化サブユニットの変動に依存しており,出芽酵母のDbf4はN末端近傍のD-box(Destruction box)を介して,G1期にユビキチンリガーゼAPC/C(anaphase promoting complex/cyclosome)依存的に分解される12, 14, 35, 36).
動物細胞のCdc7およびDbf4/ASKはいずれも休止期にはその転写が抑制されているが,血清添加等による増殖開始に伴い転写が活性化される.いずれの場合にもE2F転写因子が関与しており,休止期には抑制的なE2Fにより転写が抑制されている10).また,ヒトDbf4/ASKの増殖シグナルに応答した転写活性化には,プロモーター上の63 bpの配列のみで十分であることが示された.この配列はSp1結合部位を含み,典型的なE2F1結合部位は有しない.しかし,E2Fタンパク質の人為的発現により活性化される37).その他,ヒトDbf4/ASK遺伝子コアプロモーター領域にはMCB(MluI cell cycle box)が存在することも報告されている38).
3)Dbf4によるCdc7活性化のメカニズム(図2)
出芽酵母や分裂酵母のCdc7/Hsk1キナーゼのC末端領域は保存度が低いものの,この領域のみでDbf4活性化サブユニットに結合できることが示された39).ヒトのCdc7では二つの特有のモチーフDAM-1(Dbf4/ASK interacting motif-1;キナーゼ挿入配列IIIのN末端近くのアミノ酸448~457)およびDAM-2(C末端の10アミノ酸)が同定されており,DAM-1, DAM-2はそれぞれDbf4/ASKのDbf4-motif-MおよびDbf4-motif-Cと相互作用する32)(図2A).このようにCdc7のC末端とDbf4との相互作用は,ヒトにおいても保存されている.
ヒトDbf4/ASKはCdc7のATP結合を促進する.DAM-1を除くキナーゼ挿入配列II, IIIの大部分を欠失したCdc7はそれ自身でATP結合能と自己リン酸化能を持つが,基質のリン酸化にはDbf4との結合を必要とした.つまり,Cdc7は挿入配列によりATPの結合が阻害されており,Dbf4との結合により立体構造が変化しATPと結合できるようになる.そしてDbf4サブユニットは,効率よく基質を認識するためにも必要である32).
最近,ヒトCdc7-Dbf4/ASK複合体のX線構造解析が報告された(図2B)40).Dbf4-motif-Cを含む領域がCdc7キナーゼのN-lobeに結合し,保存されたαCヘリックスを安定化し活性化する.一方,Dbf4-motif-MはCdc7のC-lobe内のキナーゼ挿入配列IIIのDAM-1領域と相互作用する.Cdc7のDAM-2領域は構造を特定できなかったためDbf4-motif-Cとの相互作用は不明である.上述の分子解剖の結果とあわせると,Dbf4-motif-NとDbf4-motif-CがそれぞれCdc7のC-lobeとN-lobe(およびC末端尾部)に結合することで,Cdc7キナーゼを完全に活性化すると考えられる.
4. Cdc7キナーゼによる複製開始の制御と,組換え・修復・チェックポイントなどへの関与
Cdc7の機能解析は,複製開始における役割を中心に進展してきており,複製開始の制御がその中心的な機能であることに疑いはない.しかし同時に,複製以外のプロセスへの関与も,出芽酵母の変異株を用いた遺伝学的解析により,変異体が単離された早い研究の段階から示唆されていた.実際cdc7変異体では,減数分裂期の組換え効率の減少が報告され,さらに,紫外線誘導変異への影響も報告された41–44).後述のように,これらの発見の分子的基盤は30年後に解明されることになる.
本節では,これまで明らかにされているCdc7キナーゼの複製開始・そして複製チェックポイントにおける機能,そしてその後明らかになってきた組換え・修復などにおける役割と,その標的分子を紹介する(図3).
1)複製起点活性化における機能
Cdc7はG1/S期移行に必要であることは知られていたが,細胞周期の進行に必要であるのか,複製起点の活性化に必要であるのかは不明であった.1998年に出芽酵母を用いて,Cdc7は初期複製起点が活性化した後の後期複製起点の活性化にも必要であることが示され,Cdc7はS期を通じてそれぞれの複製起点の活性化に必要であることが明らかとなった45, 46).
Cdc7は複製開始の律速段階の一つとなっており,その量を増やすことにより複製起点活性化の効率を増加させることができる47).また,Cdc7を染色体上の特定の部位に人工的に局在化させると,隣接する複製起点の活性化が促進される48).これらの結果は,Cdc7キナーゼの利用効率が複製起点の活性化の頻度,効率を制御することを示す.
Cdc7のキナーゼ活性の標的(基質)は長い間不明であったが,我々のグループは,ヒトCdc7がMcmを試験管内でリン酸化することを報告し8),TyeらのグループはMcm2が重要な基質の一つであることを酵母における解析から示した49).これが契機となり,Cdc7がMcmをリン酸化することは各種生物で確認された.その後,Mcm2, Mcm4, Mcm6のN末端尾部領域がCdc7により重複的にリン酸化されることが複製開始に重要であることが示された50–52).
最近,出芽酵母のDNA複製開始反応が精製タンパク質から試験管内で再構築された.この系を用いてCdc7の複製開始に対する要求性を調べた結果,Cdc7によりリン酸化されたMcm4とMcm6がSld3を複製起点領域にリクルートすることが明らかとなった53).次いで,Sld3が複製開始の必須因子であるCdc45を複製複合体上にリクルートすることでDNA複製が開始する.
2)Cdc7の複製チェックポイントにおける機能
複製フォークの進行が何らかの障害により停止したとき,細胞は複製チェックポイントのシグナルを誘導し,高等生物ではATR→Claspin→Chk1の経路を活性化し,さらなる複製起点活性化の一時的な阻害および細胞周期進行の停止シグナルを誘起する54).Cdc7の複製チェックポイントにおける機能は,チェックポイントの標的としての機能とチェックポイントの活性化因子としての機能の二方向から研究が行われた.
カエル卵抽出液の複製系による実験から,DNA複製の阻害によりCdc7キナーゼ活性が阻害され,その結果McmヘリカーゼのコファクターであるCdc45のクロマチン結合が阻害されるという報告がなされた55).この阻害にはRPAの一本鎖領域への結合が必要とされる.出芽酵母においても,複製ストレスによりDbf4がRad53(Chk2)依存的に過剰リン酸化を受け,Cdc7–Dbf4キナーゼ活性が減少することが報告された56).これらの報告はCdc7–Dbf4キナーゼ複合体がS期チェックポイントの標的の一つであることを示す.しかしながらその後,カエル卵抽出液においてCdc7–Dbf4複合体形成,Cdc7キナーゼ活性,クロマチン結合は複製ストレスの影響を受けないことが報告され,Cdc7はチェックポイントシグナルを減弱させることにより複製の再開始を促進するというモデルが提唱された57).ヒト細胞においても複製ストレス時にCdc7キナーゼ活性は保持されているという報告がある58).分裂酵母においてもHU処理後のHsk1キナーゼ活性は変化しなかった59).したがって現状においてCdc7が複製チェックポイントの標的であるという結論はまだ議論の余地を残す.一方,出芽酵母の遺伝学的解析から,Dbf4がチェックポイントの標的の一つであることを支持する強い証拠が得られている.Dbf4上のリン酸化候補部位を網羅的に変異した結果,チェックポイントによる後期複製起点抑制が解除された.Dbf4変異と,複製開始因子Sld3上のリン酸化部位の変異を組み合わせることにより,複製阻害剤の存在下で後期複製起点が活性化され,S期へと進行した60, 61).このように,複製阻害によるチェックポイントの活性化(出芽酵母においてはRad53キナーゼの活性化)は何らかのメカニズムでDbf4のリン酸化を介して複製活性化を抑制する.しかしながら,この抑制のメカニズムは現在まで不明である.
複製因子の,複製チェックポイントの活性化因子としての機能の判断は実験的に難しい点がある.なぜなら複製ストレスチェックポイントは,進行中の複製フォークの数に依存するという報告があり62, 63),Cdc7を含め複製因子の阻害は複製そのものを阻害するため間接的な影響をみているにすぎないという可能性があるからである.しかし,出芽酵母Cdc7をバイパスできるmcm4変異体では,Cdc7を欠損させるとチェックポイント反応が誘導されなかったため,Cdc7が複製チェックポイントの活性化因子であると結論された64).同様に,分裂酵母のhsk1変異株においては,HUに応答したMrc1リン酸化,Cds1活性化が著しく減弱したが,Rad3の活性化は影響を受けなかった65).また後述のように,分裂酵母においてHsk1の要求性はrif1欠損によりバイパスされるが,この株においても複製チェックポイントは観察された(未発表データ).これらの実験結果から,Hsk1はMrc1の活性化を介してチェックポイント反応活性化に必要とされることが強く示唆された.さらに,他の複製開始因子変異体を解析した結果,McmやPolεの変異体ではCds1の活性化は強く影響されなかったが,Cdc45の変異体では強く抑制されていることが明らかとなった.Hsk1はCdc45の複製起点複合体への結合に必要とされる.これらの結果は,Hsk1がCdc45を複製起点へと誘導することとMrc1の活性化が何らかのメカニズムで連動している可能性を示唆する65).
動物細胞においてもCdc7のノックダウンは,複製ストレスによるATR活性化には影響を与えないが,酵母Mrc1の機能的ホモログであるClaspinのリン酸化を減弱させ,チェックポイント活性化を抑制する66).また,Mrc1と同様にClaspinはCdc7の基質となる66, 67).後述のようにClaspinのCdc7によるリン酸化が複製チェックポイントの活性化にも重要な役割を果たす.
3)減数分裂期の組換え
前述のように,1970年代に出芽酵母cdc7変異体の解析から,Cdc7の減数分裂期における機能が報告されていた41).cdc7変異体二倍体株の減数分裂は制限温度下ではシナプトネマ複合体形成の前で停止する.cdc7変異を許容温度に戻すと直ちにシナプトネマ複合体が形成され,組換えが進行するため,cdc7変異において減数分裂前DNA複製は進行すると推測された.一方,Mcm4をコードするcdc21あるいはDNAリガーゼをコードするcdc9変異体では,減数分裂前DNA複製が完了しなかった.
その後,Cdc7の減数分裂期における機能についての研究は進まなかったが,2000年代に入り,分裂酵母においてHsk1/Cdc7は減数分裂期組換え開始に必須な二重鎖DNA切断(double-strand break:DSB)導入に重要な役割を果たすことが示された68).また,hsk1変異株は減数分裂前DNA複製に若干の遅れがあるが,ほぼ正常に進行することからCdc7は減数分裂前DNA複製には必須でないことが示された.出芽酵母においてもATPアナログによりCdc7を特異的に不活性化できる変異体を用いた解析により,Cdc7阻害によりDNA合成は進行するが,減数分裂期組換えの直前で停止することが示された69).その後,Cdc7はMer2をリン酸化することによりDSBを制御することが示された70, 71).Cdc7は減数分裂組換え二重鎖切断誘導に必要であるMer2タンパク質上のSer30およびその他のSer/ThrをCdk(cyclin-dependent kinase)と連動してリン酸化することにより,Rec114とMei4のクロマチン結合を誘導し,その結果として,二重鎖切断を導入するSpo11のDSBホットスポットへの集合を促進する.
4)損傷乗り越えDNA修復における機能
初期に出芽酵母のcdc7変異体において観察された紫外線誘導変異42, 43)は後の研究からerror prone(間違いしがち)なTLS(translesion synthesis)ポリメラーゼによる損傷乗り越え修復に由来することが明らかとなった.動物細胞では下記の報告がなされている.複製フォーク停止によりAPC/Cの活性化因子Cdh1がChk1依存的に分解され,APC/C(Cdh1)が不活性化する.その結果,APC/C(Cdh1)の基質の一つであるCdc7-Dbf4/ASK複合体がクロマチン上で安定化する.Dbf4-motif-CはPCNAのユビキチンリガーゼRad18のN末端領域と相互作用するが,この相互作用によりRad18がクロマチン上に集合し,さらにTLSポリメラーゼであるDNAポリメラーゼ・イータ(Polη)が損傷部位にリクルートされる72).また別の報告から,Cdc7はRad18上のPolη結合部位に存在するセリン残基をリン酸化することが示された.このリン酸化に依存してPolηはRad18に結合し,複製フォーク停止部位に集合する73).
出芽酵母の解析からもCdc7はRad6経路のTLSに関与することが示された74).Cdc7はDNAポリメラーゼζ依存的な変異誘導に関与することも報告された75).
5)ヘテロクロマチン複製における機能
転写や組換えなどの反応が不活性な染色体領域であるヘテロクロマチン領域の複製は通常S期後期に起こるが,分裂酵母の代表的なヘテロクロマチン領域であるセントロメア周辺(pericentromeric)領域,および接合型(mating-type:mat)領域はS期初期に複製される.Hsk1-Dfp1/Him1複合体は,Swi6(ヘテロクロマチン形成H3K9メチル結合タンパク質HP1の分裂酵母ホモログ)のPVVTI配列(448~452残基76, 77))に依存してSwi6上にリクルートされ,この領域の初期複製を可能にしていることが示された.Dfp1/Him1をセントロメア周辺領域に人為的に結合させることにより,swi6∆細胞においてこの領域はS期初期に複製できるようになった77).
6)ヒストンの修飾によるエピゲノム制御
コアヒストンの化学修飾はエピゲノム制御の中核をなす.Cdc7はS期にヒストンH3のThr45をリン酸化することにより複製の進行と,複製ストレス応答に関与することが示された78).複製ストレス時にはヒストンH3のThr45リン酸化が蓄積するが,逆にヒストンH3 Thr45リン酸化の欠損は複製の障害を誘導した.このリン酸化は複製過程で起こり,ゲノム安定性維持のために重要であることが示されているヒストンH3のLys56のアセチル化とは独立に進行し,両者の変異は合成致死となる.
上述のように,Cdc7はMcm複合体のMcm2, 4, 6をリン酸化することが明らかとなった50, 51).in vitroのリン酸化反応では,Mcm複合体をCdkにより先にリン酸化するとCdc7によるリン酸化が大きく促進される.Mcmの主要なリン酸化部位はSer-Thr-Pro配列の最初のSer残基であり,真ん中のThr残基がCdkによりリン酸化されることにより隣接するSerがCdc7によりリン酸化されやすくなる50).Mcm以外の基質のリン酸化部位の解析からも,同様なCdkとの共同性が示されている56, 57).一般に,Cdc7のリン酸化部位は酸性アミノ酸の近傍に存在し,複数の標的部位を重複的にリン酸化する場合が多い.このような酸性領域嗜好型(acidophilic)キナーゼとしては他にカゼインキナーゼが知られている.Cdc7キナーゼの基質認識部位と考えられているキナーゼドメインVIのアミノ酸(出芽酵母ではThr167近傍,ヒトではSer181近傍)はT/Sが多く,他の多くのキナーゼとは異なっている39).出芽酵母T167E変異は,機能減弱型の表現型を示した.この部位のアミノ酸がCdc7に類似するキナーゼはカゼインキナーゼであることからもCdc7とカゼインキナーゼの基質嗜好の類似性が示唆される.
最近,Cdc7キナーゼが基質あるいはそれに付随するタンパク質に直接的にリクルートされ,効率よく基質をリン酸化する例が示された.出芽酵母では,Mcm4のN末端領域に存在するDDD配列(DDK docking domain,アミノ酸175~333)が直接Cdc7をリクルートし,その結果Mcmの効率よいリン酸化が進行する79).また出芽酵母では減数分裂期に減数分裂期前DNA複製と連動して組換えが起こるが,その際に複製フォークに存在するTof1–Csm3複合体(高等生物ではTim–Tipin複合体)にCdc7キナーゼがリクルートされ,DSB誘導に必須なMer2をリン酸化することが報告された80).
ヒト細胞では,出芽細胞Mcm4で見いだされた上記DDDに相当する領域に,Cdc7は結合しない.しかし,最近,Claspinタンパク質C末端近傍に存在する酸性アミノ酸領域(acidic patch:AP)にCdc7が特異的に結合することが明らかになった.そして,McmのCdc7によるリン酸化はこのAPの存在に依存することが示された.Mcmのリン酸化へのClaspinの要求性は正常細胞では観察されるが,Cdc7が高発現しているがん細胞では観察されない81).
このように,Cdc7が基質をリン酸化する際には,リクルータータンパク質がCdc7を基質の近くに呼び寄せて効率よくリン酸化を遂行すると考えられる.このようなリクルーターはおそらく他の基質の場合にも存在する可能性がある.
6. Cdc7の予期せぬ発見:Cdc7は複製開始に必須ではない?(表1)
表1 分裂酵母のCdc7(Hsk1)機能をバイパスする変異および生育条件 | mrc1-3A | mrc1 ∆HBS | mrc1-3A
mrc1 ∆HBS | cds1欠損 | rif1欠損 | 高温 |
|---|
| 文献83) | 文献89, 115) | 文献89) | 文献83) | 文献85, 116, 117) | 文献84) |
|---|
| Cdc7機能のバイパス | 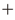 |  |  |  | 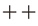 | 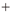 |
| 影響を受ける複製起点 | 後期起点(チェックポイント依存的) | 初期起点(チェックポイント非依存的) | 初期および後期起点 | 後期起点(チェックポイント依存的) | 後期起点(チェックポイント非依存的) | 一部の後期起点 |
| +の大きさは,バイパスの効率のよさを反映する. |
Cdc7はもともと出芽酵母の温度感受性cdc7変異株として発見されたため,複製開始・増殖に必須であるという概念が固定化されていた.したがって,我々が「分裂酵母においていろいろな変異体,あるいは増殖条件によってHsk1キナーゼは増殖に必須ではなくなる」と報告したときにはかなり懐疑的な意見をいただいた.
我々はhsk1変異とTim–Tipinの分裂酵母ホモログであるswi1–swi3変異体が合成致死になるということを先に発見していた82).Swi1–Swi3複合体とMrc1はいずれも複製フォークにおいてフォークの進行を手助けする因子であることが示唆されていたので,当然mrc1変異体とも合成致死になるのではと考え調べてみた.しかしながら,意外なことにmrc1欠損によりhsk1変異が相補された83).さらにMrc1の下流で複製チェックポイントのeffector kinaseとして機能するcds1の欠損によってもhsk1変異は弱く相補された.そこで,チェックポイント機能が不全になるとhsk1変異が相補される可能性を考慮し,mrc1のチェックポイント機能変異体3A変異(Rad3/ATRによるリン酸化部位SQ/TQをAQに変異)と掛け合わせた結果,これによってもhsk1変異が弱く相補された.さらに,予想外なことに,これらの変異は,hsk1∆株の増殖も支持した.
我々の単離したhsk1-89温度感受性変異株は30°Cでは生育できないが,37°Cでは増殖能が回復する84).さらに我々はhsk1∆株が25°C, 30°Cでは生育できないが,37°Cで生育可能であることを見いだした.分裂酵母には前述のように胞子形成時に発現するSpo4–Spo6複合体が存在し,Hsk1キナーゼの欠損を代替している可能性が残ったが,hsk1∆spo4∆二重破壊株も37°Cで生育可能であったため,この可能性は否定された.以上の発見は,分裂酵母の特殊な遺伝的背景あるいは増殖条件下ではHsk1が複製開始に必須ではないことを示している.
我々は,後期複製起点の活性化を抑制するMrc1やCds1の変異が,Hsk1機能をバイパスすることに着目し,hsk1∆株の増殖を可能にするその他の変異体を探索することにより,後期複製起点の活性化を制御する未知の因子を同定できる可能性を想定した.その結果単離された遺伝子がrif1であった84).rif1欠損株(rif1∆)はhsk1∆を強く相補し,rif1∆hsk1∆株は30°Cで効率よく増殖する.その後の研究により,Rif1は予想したように,後期複製起点活性化を抑制する複製タイミング制御因子であることを示した85).そして,Rif1による複製タイミング制御は進化的に保存されていることも証明した86, 87).Rif1はクロマチン上の特異的な部位への結合により,結合部位を囲む領域(~100 kbにわたる)の複製開始を抑制する88).すなわち,rif1∆株ではこの抑制が解除されるために,Hsk1キナーゼによるリン酸化に依存せず複製が開始できると考えられる.一方,複製チェックポイント欠損は,チェックポイントによる種々の複製抑制機構が解除されるために,複製のポテンシャルが上昇したためHsk1なしでも複製開始できるようになったと考えられた.高温の効果は不明であるが,二本鎖DNA開裂促進,クロマチン構造の弛緩などにより複製開始のポテンシャルが増加した可能性が推測できる.これらの条件下でhsk1∆が生育可能であるという事実は,複製開始にHsk1/Cdc7キナーゼは必須ではなく,効率は悪いが他のキナーゼがその役割を代替できることを示唆している.
チェックポイント特異的欠損mrc1変異(mrc1-3A)によるhsk1∆の相補は,mrc1∆による相補より効率が悪い.最近我々は,Mrc1のC末端近傍領域にHBS(Hsk1 bypass segment)と名づけた領域を同定した.このHBS領域はClaspinのAPと同様にHsk1との結合に関与する.HBSを欠損(HBS∆)すると,チェックポイント機能は正常であるが,hsk1∆を弱く相補する.mrc1-3AとHBS∆を組み合わせると,相加的に相補の効率が上昇した.この領域は複製開始にブレーキをかける機能を有しており,Hsk1キナーゼによるMrc1のリン酸化によりこのブレーキが解除される可能性が示された89).チェックポイント機能は主に後期複製起点を阻害するのに対して,HBSによるブレーキは一部の初期複製起点の発火を抑制する.
1)Cdc7の個体レベルでの機能
Cdc7欠損マウスは胎生3.5~6.5日に死亡する.またES細胞において条件的にCdc7を欠損させるとDNA複製が停止し,DNA損傷が蓄積するため,やがてp53依存的な細胞死に至る.興味深いことにp53ノックアウトマウスとCdc7ノックアウトを掛け合わせるとCdc7 p53二重欠損マウスは胎生8.5日まで生存でき,Cdc7 p53二重欠損由来のblastocysts(胚盤胞)は試験管内で内部細胞隗の形成を行うことができる.一方,MEF細胞においてCdc7欠損を誘導するとDNA複製は低下するが,細胞死は誘導されずやがて老化細胞が出現する90).
我々はまた,Cdc7活性減弱マウスを樹立した.このマウスは,誕生するが体のサイズが小さく不妊である.Cdc7遺伝子導入によるレスキュー実験ではCdc7遺伝子のコピー数を増加させることにより,精子形成は正常近くまで回復する.このように,Cdc7の発現減弱により精巣および卵巣の形成が阻害される.精子の形成は,減数分裂期初期で停止しており,酵母においてCdc7が減数分裂期組換えに必要であるという知見と合致する91).
Cdc7は神経細胞でも発現している.増殖しない細胞におけるCdc7発現の意義は不明であるが,最近,神経変性疾患をもたらすTDP-43タンパク質のSer409/410をCdc7がリン酸化することが線虫を用いて報告された92).ヒト細胞においてもこのリン酸化は観察され,前頭側頭葉変性症においてCdc7の免疫染色はリン酸化TDP-43との共局在を示す.Cdc7阻害剤によりTDP-43のリン酸化が減少し神経変性も減弱した92).今後,Cdc7の臓器特異的ノックアウトにより種々の臓器・組織の形成・機能におけるCdc7の役割を明らかにする必要がある.
2)続々明らかとなるCdc7のその他の機能
a.菌糸形成の制御
ヒトの日和見感染の原因となるカンジダ・アルビカンス(Candida albicans)のCdc7–Dbf4キナーゼ複合体は通常の増殖に必須であるが,Cdc7を欠損あるいは活性阻害を誘導すると菌糸を形成することが明らかとなった.すなわちCdc7は通常菌糸形成を阻害している93).
b.トポイソメラーゼの制御とセントロメア
ヒトDbf4/ASKはS期にセントロメアに局在し,それに引き続いてTop2Aがセントロメアにリクルートされるが,Top2Aのセントロメア局在化にはCdc7によるリン酸化が必要である.Cdc7はin vitroでTop2Aをリン酸化することができ,リン酸化部位(S1213/S1525)の変異あるいはCdc7活性の阻害によりTop2Aのセントロメアへのリクルートは遅延する94).
c.M期進行の制御
Cdc7はM期にも発現している.分裂酵母においてcdc25停止(G2/M期停止)からリリースする際にhsk1の機能が必要であることがわかった(松本ら,未発表データ).さらにヒト細胞においてもCdc7はAuroraBキナーゼの活性制御を通じてM期制御に関与する可能性が示唆されている(伊藤ら,未発表データ).
d.姉妹染色分体接着の制御
rad21+は体細胞分裂期のコヒーシン複合体のnon-SMCサブユニット(Kleisinサブユニット)をコードする.rad21-K1 hsk1-89の二重変異株は合成致死を示し95, 96),hsk1-89株は姉妹染色分体の早期分離を示すため,Hsk1は姉妹染色分体接着を制御している可能性がある95).C末端を欠損するdfp1変異体(Hsk1を完全に活性化できない)は減数分裂期のRec21の機能ホモログのRec8をリン酸化できず分解できないために減数分裂期のMII期に進行できない97).
e.減数分裂の制御
減数分裂期のMI期においては,姉妹染色分体のキネトコアが同じ紡錘極体からの微小管に結合する(monoorientation).Cdc7はmonopolin複合体のキネトコアへの局在を,monopolinサブユニットLrs4のリン酸化を介して制御するが,ここのCdc7によるLrs4のリン酸化はmonoorientationのために必須である98, 99).
f.DNA修復の制御
PCNAは,そのリング構造の中央部にDNA鎖を通して,固定され,他の重要な複製タンパク質の集合部位となることが知られており,複製クランプと呼ばれる.Rad9–Hus1–Rad1複合体は,複製ストレス時に機能するPCNAに構造が類似したクランプ(9–1–1クランプと呼ばれる)である.分裂酵母においてHsk1は,複製時に起こるDNA損傷に応答して,9–1–1クランプのRad9をリン酸化する.このリン酸化はRad4/Cut5(TopBP1)およびRad3(ATR)によるリン酸化に依存して起こり,9–1–1のRPA(replication protein A,一本鎖DNA結合タンパク質)との相互作用を減弱させることから,Rad9をDNA損傷部位から乖離させ,修復を促進させる可能性が提唱された100).
g.タンパク質分解の制御
Cdc7キナーゼによるタンパク質分解促進も報告されている.分裂酵母のS期特異的ヒストンタンパク質合成を制御するGAT A様転写因子Ams2は細胞周期特異的に合成され,その安定性はHsk1–Dfp1キナーゼによるD-boxのリン酸化に依存することが示された101).同様なHsk1キナーゼによるホスホデグロンのリン酸化を介したタンパク質分解はMrc1タンパク質においても報告されている102).
また姉妹染色分体接着の確立に関与するEco1は,Cdk1によるリン酸化の後,Cdc7–Dbf4キナーゼおよびGSK-3ホモログのMck1キナーゼにより逐次的リン酸化を受け,最終的にユビキチンリガーゼSCF(Cdc4)による分解が誘導される103).
h.クロマチン形成
ヒトCdc7はCAF1(chromatin assembly factor 1)と相互作用し,そのp150サブユニットをリン酸化する.このリン酸化はCAF1とPCNAとの相互作用を促進し,DNA複製とクロマチン形成のカップリングに貢献する104).
i.相同組換え
Cdc7とCdc5(Polo kinase)は,組換え中間体乖離因子であるMus81–Mms4を相互依存的にリン酸化する.Cdc7によるMus4のリン酸化は,分裂期におけるMus81の活性化に必要である.一方,Mus81–Mms4が結合するスキャホールドタンパク質Rtt107は,Cdc7と相互作用し,Mus81の活性化のためにリクルートする105).
正常な繊維芽細胞でCdc7をノックダウンしてもS期に進行する前に停止し細胞死に至らない.これは,p53とp21の発現が増加し,CDK活性は低下し,Rbは低リン酸化状態になり,G1期に停止するためである.このとき転写因子FoxO3aがp53を介してp21を活性化し,p15(INK4B)発現を増加させる.p53はWnt/β-cateninシグナルの拮抗因子であるDkk3を発現誘導し,MycとCycinDの発現を低下させる.Cdc7/FoxO3a, Cdc7/p15, Cdc7/p53またはCdc7/Dkk3などの二重ノックダウンを行うと,G1期停止をバイパスし,S期に進行するが,やがて細胞死により死滅する106).一方,遺伝毒性ストレス(電磁放射線,ドキソルビシン)の存在下でCdc7はp53依存的に分解され,G1期で停止する.このときp53依存的なCdc7発現抑制はmiR-192/215を介した転写後制御およびFbxw7β E3ユビキチンリガーゼを介した翻訳後制御の二つのレベルで起こる107).このように,正常細胞ではCdc7はp53と連動して,正常増殖およびDNA損傷時に細胞の生存を維持する.
これに対して,Cdc7をがん細胞においてノックダウンすると,強力な細胞死が誘導される108–110).p53を欠損したがん細胞では,CyclinB1が核内に蓄積し,やがて異常なM期に進行し細胞死する.一方p53を有するがん細胞ではCyclinB1の蓄積は観察されず異常なS期に進行し細胞死に至る111).これを利用して,Cdc7の阻害剤は,新規な制がん剤として開発が進んでいる.すでに,Cdc7のキナーゼ活性を特異的に阻害する低分子化合物が開発され,Xenograftモデルにおいてヒトがん細胞に由来する腫瘍形成を効果的に抑制する効果が報告されている112–114).
以前にHartwell博士が奥様とご一緒に来日された際,新幹線で東京から京都にお連れしたことがあった.その際,「自分はCdc7キナーゼの研究をしている」と伝えると,「cdc7は私のお気に入りの変異体の一つだ」と仰っていた.研究を開始した当時は,Cdc7は複製開始に特化したキナーゼと考えられていたが,その詳細は作用機序も含めてまったく不明であった.また他の生物種における存在も知られていなかった.25年経過して,Cdc7は他の多くの細胞周期制御因子と同様に酵母からヒトまで機能の保存された分子であることが明らかになり,さらに,複製制御におけるCdc7の機能の大枠は明らかになった.今後in vitro複製系を用いて,その詳細なメカニズムが解明されるのも時間の問題であろう.
しかしながら,最も意外な展開の一つは,Cdc7の複製以外の種々の生体制御への関与であろう.現在ではCdc7は複製キナーゼであるとともに,生体反応のmodulator kinaseといってもよいかと思われる.Cdc7による酸性領域の認識とそれによる複数のセリン・トレオニン残基のリン酸化,さらにそれに引き続く他の因子によるリン酸化領域の認識,あるいはリン酸化による構造変化について,より一般的な分子基盤の解明が今後の課題である.
もう一つの意外な発見は,Cdc7は効率よい複製開始に必要とされるが,Cdc7なしで複製を開始しうる経路が種々の生物で存在することである.マウス個体においても,Cdc7の要求性は種々の細胞,組織,臓器などにより異なるようである(筆者らの未発表データ).Cdc7キナーゼの要求性のありなしが,複製開始の様式にどのような影響を及ぼすかは明らかでなく,複製開始様式の多様性を考慮する上でも興味深い.
謝辞Acknowledgments
本総説で紹介した研究成果のなかで,我々の研究室によるものは,これまでの多くの共同研究者の努力の賜物であります.私が1990年に留学から帰国し,Cdc7の研究を開始してから現在に至るまでのすべての共同研究者の皆様に感謝いたします.また,大学院の一年時から私をDNA複製の研究にいざない,研究を一からご指導いただいた新井賢一先生には,あらためてここに深い感謝の意を表します.また,2000年に東京都医学総合研究所の前身の東京都臨床医学総合研究所に研究室を構えて以来,田中啓二先生,米川博通先生,宇井理生先生にはひとかたならぬご支援とご指導をいただきましたことをここに心から感謝いたします.
引用文献References
1) Hartwell, L.H., Culotti, J., & Reid, B. (1970) Proc. Natl. Acad. Sci. USA, 66, 352–359.
2) Hartwell, L.H. (1973) J. Bacteriol., 115, 966–974.
3) Patterson, M., Sclafani, R.A., Fangman, W.L., & Rosamond, J. (1986) Mol. Cell. Biol., 6, 1590–1598.
4) Johnston, L.H. & Thomas, A.P. (1982) Mol. Gen. Genet., 186, 445–448.
5) Jackson, A.L., Pahl, P.M., Harrison, K., Rosamond, J., & Sclafani, R.A. (1993) Mol. Cell. Biol., 13, 2899–2908.
6) Kitada, K., Johnston, L.H., Sugino, T., & Sugino, A. (1992) Genetics, 131, 21–29.
7) Masai, H., Miyake, T., & Arai, K. (1995) EMBO J., 14, 3094–3104.
8) Sato, N., Arai, K., & Masai, H. (1997) EMBO J., 16, 4340–4351.
9) Jiang, W. & Hunter, T. (1997) Proc. Natl. Acad. Sci. USA, 94, 14320–14325.
10) Kim, J.M., Sato, N., Yamada, M., Arai, K., & Masai, H. (1998) J. Biol. Chem., 273, 23248–23257.
11) Brown, G.W. & Kelly, T.J. (1998) J. Biol. Chem., 273, 22083–22090.
12) Takeda, T., Ogino, K., Matsui, E., Cho, M.K., Kumagai, H., Miyake, T., Arai, K., & Masai, H. (1999) Mol. Cell. Biol., 19, 5535–5547.
13) Brown, G.W. & Kelly, T.J. (1999) Proc. Natl. Acad. Sci. USA, 96, 8443–8448.
14) Kumagai, H., Sato, N., Yamada, M., Mahony, D., Seghezzi, W., Lees, E., Arai, K., & Masai, H. (1999) Mol. Cell. Biol., 19, 5083–5095.
15) Jiang, W., McDonald, D., Hope, T.J., & Hunter, T. (1999) EMBO J., 18, 5703–5713.
16) Nakamura, T., Nakamura-Kubo, M., Nakamura, T., & Shimoda, C. (2002) Mol. Cell. Biol., 22, 309–320.
17) Montagnoli, A., Bosotti, R., Villa, F., Rialland, M., Brotherton, D., Mercurio, C., Berthelsen, J., & Santocanale, C. (2002) EMBO J., 21, 3171–3181.
18) Yanow, S.K., Gold, D.A., Yoo, H.Y., & Dunphy, W.G. (2003) J. Biol. Chem., 278, 41083–41092.
19) Yoshizawa-Sugata, N., Ishii, A., Taniyama, C., Matsui, E., Arai, K., & Masai, H. (2005) J. Biol. Chem., 280, 13062–13070.
20) Takahashi, T.S. & Walter, J.C. (2005) Genes Dev., 19, 2295–2300.
21) Matsumoto, S. & Masai, H. (2013) Biochem. Soc. Trans., 41, 1712–1719.
22) Matthews, L.A. & Guarné, A. (2013) Cell Cycle, 12, 1180–1188.
23) Masai, H. & Arai, K. (2000) Biochem. Biophys. Res. Commun., 275, 228–232.
24) Ogino, K., Takeda, T., Matsui, E., Iiyama, H., Taniyama, C., Arai, K., & Masai, H. (2001) J. Biol. Chem., 276, 31376–31387.
25) Varrin, A.E., Prasad, A.A., Scholz, R.P., Ramer, M.D., & Duncker, B.P. (2005) Mol. Cell. Biol., 25, 7494–7504.
26) Chen, Y.C., Kenworthy, J., Gabrielse, C., Hänni, C., Zegerman, P., & Weinreich, M. (2013) Genetics, 194, 389–401.
27) Matthews, L.A., Selvaratnam, R., Jones, D.R., Akimoto, M., McConkey, B.J., Melacini, G., Duncker, B.P., & Guarné, A. (2014) J. Biol. Chem., 289, 2589–2599.
28) Harkins, V., Gabrielse, C., Haste, L., & Weinreich, M. (2009) Genetics, 183, 1269–1282.
29) Fung, A.D., Ou, J., Bueler, S., & Brown, G.W. (2002) Mol. Cell. Biol., 22, 4477–4490.
30) Jones, D.R., Prasad, A.A., Chan, P.K., & Duncker, B.P. (2010) Cell Cycle, 9, 2018–2026.
31) Yamashita, N., Kim, J.M., Koiwai, O., Arai, K., & Masai, H. (2005) Genes Cells, 10, 551–563.
32) Kitamura, R., Fukatsu, R., Kakusho, N., Cho, Y.S., Taniyama, C., Yamazaki, S., Toh, G.T., Yanagi, K., Arai, N., Chang, H.J., & Masai, H. (2011) J. Biol. Chem., 286, 23031–23043.
33) Hughes, S., Jenkins, V., Dar, M.J., Engelman, A., & Cherepanov, P. (2010) J. Biol. Chem., 285, 541–554.
34) Chapman, J.W. & Johnston, L.H. (1989) Exp. Cell Res., 180, 419–428.
35) Oshiro, G., Owens, J.C., Shellman, Y., Sclafani, R.A., & Li, J.J. (1999) Mol. Cell. Biol., 19, 4888–4896.
36) Ferreira, M.F., Santocanale, C., Drury, L.S., & Diffley, J.F. (2000) Mol. Cell. Biol., 20, 242–248.
37) Yamada, M., Sato, N., Taniyama, C., Ohtani, K., Arai, K., & Masai, H. (2002) J. Biol. Chem., 277, 27668–27681.
38) Wu, X. & Lee, H. (2002) Oncogene, 21, 7786–7796.
39) Ohtoshi, A., Miyake, T., Arai, K., & Masai, H. (1997) Mol. Gen. Genet., 254, 562–570.
40) Hughes, S., Elustondo, F., Di Fonzo, A., Leroux, F.G., Wong, A.C., Snijders, A.P., Matthews, S.J., & Cherepanov, P. (2012) Nat. Struct. Mol. Biol., 19, 1101–1107.
41) Schild, D. & Byers, B. (1978) Chromosoma, 70, 109–130.
42) Njagi, G.D. & Kilbey, B.J. (1982) Mol. Gen. Genet., 186, 478–481.
43) Njagi, G.D. & Kilbey, B.J. (1982) Mutat. Res., 105, 313–318.
44) Kilbey, B.J. (1986) Mutagenesis, 1, 29–31.
45) Bousset, K. & Diffley, J.F. (1998) Genes Dev., 12, 480–490.
46) Donaldson, A.D., Fangman, W.L., & Brewer, B.J. (1998) Genes Dev., 12, 491–501.
47) Wu, P.Y. & Nurse, P. (2009) Cell, 136, 852–864.
48) Patel, P.K., Kommajosyula, N., Rosebrock, A., Bensimon, A., Leatherwood, J., Bechhoefer, J., & Rhind, N. (2008) Mol. Biol. Cell, 19, 5550–5558.
49) Lei, M., Kawasaki, Y., Young, M.R., Kihara, M., Sugino, A., & Tye, B.K. (1997) Genes Dev., 11, 3365–3374.
50) Masai, H., Matsui, E., You, Z., Ishimi, Y., Tamai, K., & Arai, K. (2000) J. Biol. Chem., 275, 29042–29052.
51) Masai, H., Taniyama, C., Ogino, K., Matsui, E., Kakusho, N., Matsumoto, S., Kim, J.M., Ishii, A., Tanaka, T., Kobayashi, T., Tamai, K., Ohtani, K., & Arai, K. (2006) J. Biol. Chem., 281, 39249–39261.
52) Montagnoli, A., Valsasina, B., Brotherton, D., Troiani, S., Rainoldi, S., Tenca, P., Molinari, A., & Santocanale, C. (2006) J. Biol. Chem., 281, 10281–10290.
53) Deegan, T.D., Yeeles, J.T., & Diffley, J.F. (2016) EMBO J., 35, 961–973.
54) Masai, H., Matsumoto, S., You, Z., Yoshizawa-Sugata, N., & Oda, M. (2010) Annu. Rev. Biochem., 79, 89–130.
55) Costanzo, V., Shechter, D., Lupardus, P.J., Cimprich, K.A., Gottesman, M., & Gautier, J. (2003) Mol. Cell, 11, 203–213.
56) Weinreich, M. & Stillman, B. (1999) EMBO J., 18, 5334–5346.
57) Tsuji, T., Lau, E., Chiang, G.G., & Jiang, W. (2008) Mol. Cell, 32, 862–869.
58) Tenca, P., Brotherton, D., Montagnoli, A., Rainoldi, S., Albanese, C., & Santocanale, C. (2007) J. Biol. Chem., 282, 208–215.
59) Matsumoto, S., Shimmoto, M., Kakusho, N., Yokoyama, M., Kanoh, Y., Hayano, M., Russell, P., & Masai, H. (2010) Cell Cycle, 9, 4627–4637.
60) Lopez-Mosqueda, J., Maas, N.L., Jonsson, Z.O., Defazio-Eli, L.G., Wohlschlegel, J., & Toczyski, D.P. (2010) Nature, 467, 479–483.
61) Zegerman, P. & Diffley, J.F. (2010) Nature, 467, 474–478.
62) Shimada, K., Pasero, P., & Gasser, S.M. (2002) Genes Dev., 16, 3236–3252.
63) Tercero, J.A., Longhese, M.P., & Diffley, J.F. (2003) Mol. Cell, 11, 1323–1336.
64) Sheu, Y.J. & Stillman, B. (2010) Nature, 463, 113–117.
65) Matsumoto, S., Shimmoto, M., Kakusho, N., Yokoyama, M., Kanoh, Y., Hayano, M., Russell, P., & Masai, H. (2010) Cell Cycle, 9, 4627–4637.
66) Kim, J.M., Kakusho, N., Yamada, M., Kanoh, Y., Takemoto, N., & Masai, H. (2008) Oncogene, 27, 3475–3482.
67) Rainey, M.D., Harhen, B., Wang, G.N., Murphy, P.V., & Santocanale, C. (2013) Cell Cycle, 12, 1560–1568.
68) Ogino, K., Hirota, K., Matsumoto, S., Takeda, T., Ohta, K., Arai, K., & Masai, H. (2006) Proc. Natl. Acad. Sci. USA, 103, 8131–8136.
69) Wan, L., Zhang, C., Shokat, K.M., & Hollingsworth, N.M. (2006) Genetics, 174, 1767–1774.
70) Sasanuma, H., Hirota, K., Fukuda, T., Kakusho, N., Kugou, K., Kawasaki, Y., Shibata, T., Masai, H., & Ohta, K. (2008) Genes Dev., 22, 398–410.
71) Wan, L., Niu, H., Futcher, B., Zhang, C., Shokat, K.M., Boulton, S.J., & Hollingsworth, N.M. (2008) Genes Dev., 22, 386–397.
72) Yamada, M., Watanabe, K., Mistrik, M., Vesela, E., Protivankova, I., Mailand, N., Lee, M., Masai, H., Lukas, J., & Bartek, J. (2013) Genes Dev., 27, 2459–2472.
73) Day, T.A., Palle, K., Barkley, L.R., Kakusho, N., Zou, Y., Tateishi, S., Verreault, A., Masai, H., & Vaziri, C. (2010) J. Cell Biol., 191, 953–966.
74) Pessoa-Brandão, L. & Sclafani, R.A. (2004) Genetics, 167, 1597–1610.
75) Brandão, L.N., Ferguson, R., Santoro, I., Jinks-Robertson, S., & Sclafani, R.A. (2014) Genetics, 197, 1111–1122.
76) Bailis, J.M., Bernard, P., Antonelli, R., Allshire, R.C., & Forsburg, S.L. (2003) Nat. Cell Biol., 5, 1111–1116.
77) Hayashi, M.T., Takahashi, T.S., Nakagawa, T., Nakayama, J., & Masukata, H. (2009) Nat. Cell Biol., 11, 357–362.
78) Baker, S.P., Phillips, J., Anderson, S., Qiu, Q., Shabanowitz, J., Smith, M.M., Yates, J.R. 3rd, Hunt, D.F., & Grant, P.A. (2010) Nat. Cell Biol., 12, 294–298.
79) Sheu, Y.J. & Stillman, B. (2006) Mol. Cell, 24, 101–113.
80) Murakami, H. & Keeney, S. (2014) Cell, 158, 861–873.
81) Yang, C.C., Suzuki, M., Yamakawa, S., Uno, S., Ishii, A., Yamazaki, S., Fukatsu, R., Fujisawa, R., Sakimura, K., Tsurimoto, T., & Masai, H. (2016) Nat. Commun., 7, 12135.
82) Matsumoto, S., Ogino, K., Noguchi, E., Russell, P., & Masai, H. (2005) J. Biol. Chem., 280, 42536–42542.
83) Hayano, M., Kanoh, Y., Matsumoto, S., & Masai, H. (2011) Mol. Cell. Biol., 31, 2380–2391.
84) Matsumoto, S., Hayano, M., Kanoh, Y., & Masai, H. (2011) J. Cell Biol., 195, 387–401.
85) Hayano, M., Kanoh, Y., Matsumoto, S., Renard-Guillet, C., Shirahige, K., & Masai, H. (2012) Genes Dev., 26, 137–150.
86) Yamazaki, S., Ishii, A., Kanoh, Y., Oda, M., Nishito, Y., & Masai, H. (2012) EMBO J., 31, 3667–3677.
87) Cornacchia, D., Dileep, V., Quivy, J.P., Foti, R., Tili, F., Santarella-Mellwig, R., Antony, C., Almouzni, G., Gilbert, D.M., & Buonomo, S.B. (2012) EMBO J., 31, 3678–3690.
88) Kanoh, Y., Matsumoto, S., Fukatsu, R., Kakusho, N., Kono, N., Renard-Guillet, C., Masuda, K., Iida, K., Nagasawa, K., Shirahige, K., & Masai, H. (2015) Nat. Struct. Mol. Biol., 22, 889–897.
89) Matsumoto, S., Kanoh, Y., Shimmoto, M., Hayano, M., Ueda, K., Fukatsu, R., Kakusho, N., & Masai, H. (2017) Mol. Cell. Biol., 37, e00355-16.
90) Kim, J.M., Nakao, K., Nakamura, K., Saito, I., Katsuki, M., Arai, K., & Masai, H. (2002) EMBO J., 21, 2168–2179.
91) Kim, J.M., Takemoto, N., Arai, K., & Masai, H. (2003) EMBO J., 22, 5260–5272.
92) Liachko, N.F., McMillan, P.J., Guthrie, C.R., Bird, T.D., Leverenz, J.B., & Kraemer, B.C. (2013) Ann. Neurol., 74, 39–52.
93) Lai, W.C., Chang, T.W., Wu, C.H., Yang, S.Y., Lee, T.L., Li, W.C., Chien, T., Cheng, Y.C., & Shieh, J.C. (2016) Sci. Rep., 6, 33716.
94) Wu, K.Z., Wang, G.N., Fitzgerald, J., Quachthithu, H., Rainey, M.D., Cattaneo, A., Bachi, A., & Santocanale, C. (2016) Nucleic Acids Res., 44, 8786–8798.
95) Takeda, T., Ogino, K., Tatebayashi, K., Ikeda, H., Arai, K., & Masai, H. (2001) Mol. Biol. Cell, 12, 1257–1274.
96) Snaith, H.A., Brown, G.W., & Forsburg, S.L. (2000) Mol. Cell. Biol., 20, 7922–7932.
97) Le, A.H., Mastro, T.L., & Forsburg, S.L. (2013) Biol. Open, 2, 728–738.
98) Lo, H.C., Wan, L., Rosebrock, A., Futcher, B., & Hollingsworth, N.M. (2008) Mol. Biol. Cell, 19, 4956–4967.
99) Matos, J., Lipp, J.J., Bogdanova, A., Guillot, S., Okaz, E., Junqueira, M., Shevchenko, A., & Zachariae, W. (2008) Cell, 135, 662–678.
100) Furuya, K., Miyabe, I., Tsutsui, Y., Paderi, F., Kakusho, N., Masai, H., Niki, H., & Carr, A.M. (2010) Mol. Cell, 40, 606–618.
101) Takayama, Y. & Toda, T. (2010) Cell Div., 5, 18.
102) Shimmoto, M., Matsumoto, S., Odagiri, Y., Noguchi, E., Russell, P., & Masai, H. (2009) Genes Cells, 14, 669–682.
103) Lyons, N.A., Fonslow, B.R., Diedrich, J.K., Yates, J.R. 3rd, & Morgan, D.O. (2013) Nat. Struct. Mol. Biol., 20, 194–201.
104) Gérard, A., Koundrioukoff, S., Ramillon, V., Sergère, J.C., Mailand, N., Quivy, J.P., & Almouzni, G. (2006) EMBO Rep., 7, 817–823.
105) Princz, L.N., Wild, P., Bittmann, J., Aguado, F.J., Blanco, M.G., Matos, J., & Pfander, B. (2017) EMBO J., 36, 664–678.
106) Tudzarova, S., Trotter, M.W., Wollenschlaeger, A., Mulvey, C., Godovac-Zimmermann, J., Williams, G.H., & Stoeber, K. (2010) EMBO J., 29, 3381–3394.
107) Tudzarova, S., Mulholland, P., Dey, A., Stoeber, K., Okorokov, A.L., & Williams, G.H. (2016) Cell Cycle, 15, 2958–2972.
108) Ito, S., Taniyami, C., Arai, N., & Masai, H. (2008) Drug News Perspect., 21, 481–488.
109) Sawa, M. & Masai, H. (2009) Drug Des. Devel. Ther., 2, 255–264.
110) Rodriguez-Acebes, S., Proctor, I., Loddo, M., Wollenschlaeger, A., Rashid, M., Falzon, M., Prevost, A.T., Sainsbury, R., Stoeber, K., & Williams, G.H. (2010) Am. J. Pathol., 177, 2034–2045.
111) Ito, S., Ishii, A., Kakusho, N., Taniyama, C., Yamazaki, S., Fukatsu, R., Sakaue-Sawano, A., Miyawaki, A., & Masai, H. (2012) PLoS One, 7, e36372.
112) Vanotti, E., Amici, R., Bargiotti, A., Berthelsen, J., Bosotti, R., Ciavolella, A., Cirla, A., Cristiani, C., D’Alessio, R., Forte, B., Isacchi, A., Martina, K., Menichincheri, M., Molinari, A., Montagnoli, A., Orsini, P., Pillan, A., Roletto, F., Scolaro, A., Tibolla, M., Valsasina, B., Varasi, M., Volpi, D., & Santocanale, C. (2008) J. Med. Chem., 51, 487–501.
113) Montagnoli, A., Valsasina, B., Croci, V., Menichincheri, M., Rainoldi, S., Marchesi, V., Tibolla, M., Tenca, P., Brotherton, D., Albanese, C., Patton, V., Alzani, R., Ciavolella, A., Sola, F., Molinari, A., Volpi, D., Avanzi, N., Fiorentini, F., Cattoni, M., Healy, S., Ballinari, D., Pesenti, E., Isacchi, A., Moll, J., Bensimon, A., Vanotti, E., & Santocanale, C. (2008) Nat. Chem. Biol., 4, 357–365.
114) Irie, T., Asami, T., Sawa, A., Uno, Y., Hanada, M., Taniyama, C., Funakoshi, Y., Masai, H., & Sawa, M. (2017) Eur. J. Med. Chem., 130, 406–418.
115) Masai, H., Yang, C.C., & Matsumoto, S. (2017) Curr. Genet., doi: 10.1007/s00294-017-0690-y.
116) Yamazaki, S., Hayano, M., & Masai, H. (2013) Trends Genet., 29, 449–460.
117) Masai, H., Kanoh, Y., Moriyama, K., Yamazaki, S., Yoshizawa, N., & Matsumoto, S. (2017) Genes Genet. Syst., doi: 10.1266/ggs.17-00008.
著者紹介Author Profile
 正井 久雄( まさい ひさお)
正井 久雄( まさい ひさお)東京都医学総合研究所副所長.理学博士.
略歴1959年愛知県に生る.81年東京大学理学部卒業.90年東京大学医科学研究所助手.95年同助教授.2000年東京都臨床医学総合研究所室長.15年より現職.
研究テーマと抱負大学院の一年からこれまでDNA複製の研究に携わってきた.その過程で,大腸菌から真核細胞に至るまで,DNAが作る特殊構造が重要な機能を果たすことを見出してきた.種を超えて保存されるDNA複製の普遍的な機構を明らかにしたい.
ウェブサイトhttp://www.igakuken.or.jp/genome/
趣味音楽,旅行先でのジョギング.