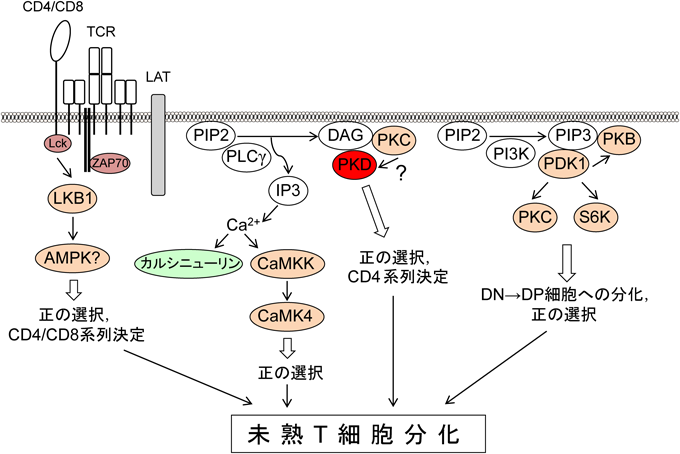
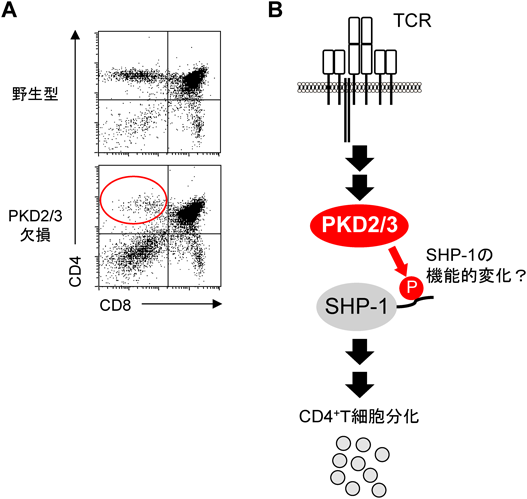
プロテインキナーゼDによる未熟T細胞の分化制御The role of serine/threonine kinase, protein kinase D, in thymocyte development
1 大阪大学微生物病研究所分子免疫制御分野Department of Molecular Immunology, Research Institute for Microbial Diseases, Osaka University ◇ 〒565–0871 大阪府吹田市山田丘3–1 ◇ 3–1 Yamadaoka, Suita, Osaka 565–0871, Japan
2 大阪大学免疫学フロンティア研究センター分子免疫学Laboratory of Molecular Immunology, Immunology Frontier Research Center, Osaka University ◇ 〒565–0871 大阪府吹田市山田丘3–1 ◇ 3–1 Yamadaoka, Suita, Osaka 565–0871, Japan
3 九州大学生体防御医学研究所分子免疫学分野Division of Molecular Immunology, Medical Institute of Bioregulation, Kyushu University ◇ 〒812–8582 福岡県福岡市東区馬出3–1–1 ◇ 3–1–1 Maidashi, Higashi-ku, Fukuoka 812–8582, Japan
発行日:2017年10月25日Published: October 25, 2017