細胞内で合成・蓄積された極性分子は,脂質二重層で構成された細胞膜を通過することができないため,いったん膜で囲まれた小胞に含まれた後,小胞膜と細胞膜が融合することにより,細胞外に放出される.この過程をエキソサイトーシス(開口放出)と呼ぶが,単に小胞内容物を細胞外に放出するだけではなく,小胞膜上のタンパク質や脂質を細胞膜に供給する役割も担う.分泌経路に乗るタンパク質や脂質は,まず,小胞体で合成され,ゴルジ体を経由し(初期分泌経路ともいわれる),トランスゴルジ網で種々の小胞に選別されて目的地に輸送される.このうち細胞分裂・分化,エネルギー摂取・消費など,比較的長期にわたる過程を制御する分子は,生合成に応じて,トランスゴルジ網より出芽する不定形の管状構造体により,間断なく細胞膜に向かって送り出される.出芽した管状小胞は,微小管に沿って延長・移動し,細胞膜に至ると融合する.この構成性分泌経路の律速段階が数時間かかる生合成過程であるのに対して,調節性分泌経路では,あらかじめ合成された生理活性物質を細胞内の小胞に貯蔵し,細胞外刺激に応じて開口放出する.その律速段階は,分泌小胞の開口放出過程であるため,外界からの刺激に対してミリ秒から分単位の時間で応答できる.その応答性の速度から,調節性分泌機構が,特に多細胞生物の細胞間機能調節に重要であることは,神経細胞,内・外分泌細胞,免疫・血球系細胞などの働きを考えれば明らかであろう.調節性分泌経路における分泌小胞は,その生成過程および形態学的特徴により,シナプス(様)小胞,有芯顆粒(分泌顆粒),リソソーム様オルガネラなどに分類される.グルタミン酸,アセチルコリンなどの低分子神経伝達物質はシナプス間隙からの再吸収や細胞質での合成により再充填されるため,シナプス小胞は,軸索先端終末部でエキソサイトーシスとエンドサイトーシス(細胞膜が陥入してくびり取られ,小胞が形成されることにより,細胞外成分や細胞膜上のタンパク質や脂質を細胞内に取り込む過程を指す)を繰り返す.一方,内・外分泌細胞のペプチドホルモンや消化酵素などのタンパク質は初期分泌経路で新たに生成されなければならず,分泌顆粒は,トランスゴルジ網を経由して出芽する.特定の刺激に応じてメラニン,界面活性剤,凝固因子などを細胞外に放出する,色素細胞メラノソーム,II型肺上皮細胞層状体,血球系細胞顆粒様オルガネラなどは,リソソーム様オルガネラと総称される.これらは,膜上に単量体GTPaseのRab27が局在する酸性オルガネラで,分泌顆粒と共通の特徴を有するが,その内容物および膜成分の少なくとも一部は,直接,トランスゴルジ網に由来するのではなく,エンドソーム系を介して生成される.このように分泌小胞の種類により,その生成や輸送経路が異なり,トランスゴルジ網以降の過程に多様な特異的分子が機能していると考えられる.しかし,調節性分泌経路の共通する特徴は,外界刺激依存性に小胞膜と細胞膜の融合が起こる最終過程にあるため,この点に着目した研究が中心となって行われてきた.すなわち,刺激を加えると,通常,一部の分泌小胞のみが開口放出するが,細胞内のどこに局在し,どのような機能的特徴を持つ小胞が開口放出可能なのか,また,多くの場合,刺激後の細胞内Ca2+濃度上昇を引き金にして分泌小胞膜と細胞膜の融合が起こるが,Ca2+を感知する機構は何か,といった課題である.
調節性分泌経路のうち,神経細胞シナプス小胞内の伝達物資は,刺激後ミリ秒以下の単位でシナプス間隙内に放出され,最速の分泌反応を示す.そのため,アクティヴ・ゾーン内の細胞膜にあらかじめ接着した小胞が,刺激後,即座に開口放出されると考えられた.また,副腎髄質や膵島の内分泌細胞においても,電子顕微鏡下,確かに細胞膜に接着している分泌顆粒が存在することが観察された.分泌小胞が定常的に細胞膜に接着している状態をドッキング(docking)と呼ぶ.他の細胞内膜輸送経路においても,ドナー膜から生成された小胞膜を正しいターゲット膜につなぎ止める機構,繋留[テザリング(tethering)]という過程が存在するが,繋留された小胞膜は,通常,構成的に膜融合を起こすため,ターゲット膜に接着したドッキングの状態で安定的に滞っていることはない.たとえば,小胞体から生成されるCOPII小胞や,構成性分泌経路の小胞が,標的膜に接着した像を電子顕微鏡写真上で捕らえることはまれである.これに対して,調節性分泌経路では,定常的に細胞膜に繋留された状態で滞っている小胞が存在する.一般に,内分泌細胞でみられる,細胞膜に接着したドッキング顆粒の数は,刺激によって開口放出される顆粒の数よりも多いことから,ドッキング顆粒の一部のみ,何らかの修飾反応,プライミング(priming)を受けて開口放出可能となっていると想定された.そこで一般に,調節性分泌経路の最終開口放出過程は,小胞の細胞膜ドッキング→プライミング→Ca2+感知→膜融合という逐次連続過程よりなると考えられている.最後の膜融合過程は,他の膜輸送経路同様,小胞膜側と細胞膜側にあるSNAREタンパク質の結合によることは確立しているが,その前段階の分子機構は十分には解明されていない.たとえば,分泌小胞の種類により,刺激後,最終的な開口放出に至るまでの時間は大きく異なることから1),Ca2+上昇によって誘導されるステップが必ずしも最終的な膜融合反応のみであるとは限らない.また,ドッキングが電子顕微鏡上の形態学的概念であるのに対し,プライミングは機能的概念であるため,両者の関係を実験により実証することは必ずしも容易ではない.さらに,後述するように,ドッキングやプライミングの定義が研究者や実験手法により異なり,これらの用語が必ずしも同一の生物事象に対して用いられておらず,混乱が生じている.このような背景のもと,本稿では,筆者の主たる研究対象である,内分泌細胞の有芯顆粒,特に膵β細胞のインスリン顆粒が,細胞膜直下に局在し,最終的な膜融合に至る過程について概説したい.筆者は,単量体GTPase Rab27とそのエフェクターが調節性分泌経路に特異的に機能していることを発見し,長年,研究を続けてきた2, 3).そのため,この見地からの記述が多くなることをご容赦願いたい.
2. ドッキングにおける膜融合装置SNAREの役割
膜融合装置SNAREタンパク質は,細胞内膜の種類により異なる分子が局在することから,当初,膜融合反応を引き起こすのみならず,融合を起こす膜の組合わせ,すなわち膜融合の特異性を決めていると考えられた.そこで,分泌小胞の細胞膜へのドッキングは,小胞膜側のv(esicle)-SNAREと細胞膜側のt(arget)-SNAREが結合することによって起こると想定された.しかし,神経細胞や副腎髄質細胞で働く,v-SNAREのSynaptobrevin 2(別名VAMP2)4, 5),t-SNAREのSNAP-256),Syntaxin 14, 7, 8)などの分子を遺伝子改変や神経毒素による切断で欠失させても,分泌すなわち膜融合反応は阻害されるものの,シナプス小胞や分泌顆粒のドッキングには影響がないことが判明した.その後,Syntaxin 1~3およびSNAP-25をすべて切断しうるボツリヌス菌毒素BONT/Cで処理した副腎髄質細胞や9),遺伝子工学によりSyntaxin 1を欠損させた膵β細胞で10),分泌顆粒のドッキング障害が報告された.しかし,筆者らがSyntaxin 1欠損膵β細胞を用いた研究では,後述するGranuphilin欠損細胞に認められるような,明瞭な細胞膜直下の分泌顆粒の減少が認められなかった11).また,SNAP-25欠損副腎髄質細胞においてもドッキング障害が報告されたが12),同一の研究室が作製したノックアウト・マウス由来の同細胞を用いた以前の研究では,ドッキングに影響がないと報告されている6).
このような不一致が生じる一因として,ドッキングの定義の問題があげられる.ドッキングは,分泌小胞膜と細胞膜の接着という,一見,明確な電子顕微鏡写真上の形態学的概念であるが,たとえば,通常用いられる50~100 nmの超薄切片内に,直径300~350 nmの分泌顆粒の正中線が入っているとは限らず,一つ一つの分泌小胞の接着の有無を判定することは必ずしも容易ではない.したがって,小胞膜あるいは小胞中心と,細胞膜の距離を計測し,一定距離以下のものをドッキング小胞とし,その数あるいは密度を比較することが多い.そのため,電子顕微鏡写真の分解能や,小胞の位置に関する基準設定の違いにより,異なる結論が導かれうる.また,通常の電子顕微鏡標本作製に用いられる化学固定や脱水処理が,標本の収縮や膜の変形を引き起こし,分泌小胞のドッキングに影響を及ぼす可能性がある.実際,高圧凍結法で試料を作製すると,Syntaxinを欠損する線虫の神経細胞や13),Synaptobrevin 2を欠損させたPC12細胞株14)において,細胞膜近傍の小胞の分布に差を見いだせるようになるという.すなわち,SNARE分子欠損細胞の高圧凍結標本では,通常の化学固定標本では必ずしも認められない,細胞膜にきわめて近接した小胞の減少が認められ,SNARE複合体の形成不全を反映していると考えられる.一方,後に述べるように,Munc18-115)やGranuphilin16)を欠損する細胞では,従来の化学固定標本において分泌顆粒のドッキング障害がはっきりと認められ,これは,SNARE複合体形成より前の,小胞の細胞膜へのデザリングの異常を示していると考えられる.したがって,各研究で,電子顕微鏡標本の作製法や,ドッキングの定義により,異なる過程を解析している可能性があり,注意を要する(9節に後述).
3. 分泌顆粒のドッキング装置Granuphilin
筆者らは,膵β細胞や下垂体内分泌細胞に特異的に高発現するGranuphilinという分子を発見し17),本分子が,分泌顆粒膜上に存在するRab27aまたはRab27bにGTP依存性に結合するエフェクターであること18, 19),細胞膜上のリン脂質やSyntaxin 1に結合活性を有し17, 20),内分泌細胞株に過剰発現すると,分泌顆粒を細胞膜近傍に集積させ21),同時にホルモン分泌反応を抑制すること20)を見いだした.本分子を欠損するマウスを作製し,その膵β細胞や下垂体細胞を,通常の電子顕微鏡化学固定標本で観察したところ,野生型細胞で認められる,細胞膜に接着した分泌顆粒がほぼ消失していることを見いだした16, 22).また,Granuphilin欠損膵β細胞では,著明なドッキング障害があるにもかかわらず,刺激依存性インスリン分泌が増大することが判明した.膵β細胞では,グルコース刺激後,2~3分以内にインスリン分泌がピークとなり,それに引き続いて,より低いレベルで持続的な分泌反応が起こる,二相性の分泌パターンを示すが,Granuphilin欠損細胞は,第一相,第二相とも野生型細胞の2倍程度の分泌量の増大を示した.また,ドッキング顆粒の消失は,細胞を刺激のない状態に維持しても認められたことから,開口放出増大の結果,細胞膜近傍の顆粒数が減少しているわけではなかった.これらの知見は,これまでの定説と異なり,分泌顆粒のドッキングは,開口放出に必須の前過程ではなく,むしろこれを抑制することを強く示唆した.
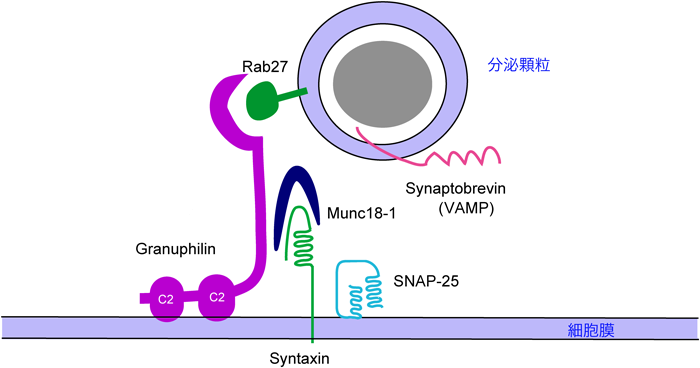
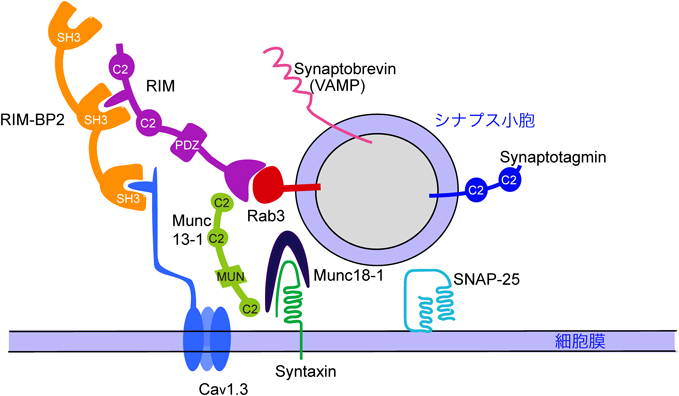
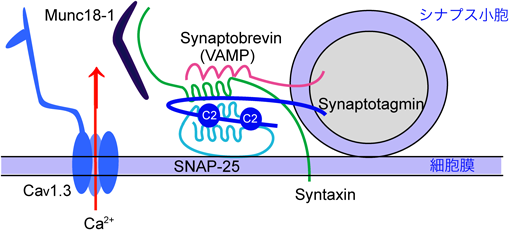
細胞内膜輸送をつかさどるRabエフェクターの最も一般的な作用は,小胞膜を標的膜に繋留させることで23),Rab27エフェクターGranuphilinのドッキング作用は,他の膜輸送経路におけるテザリングに相当すると考えらえる.ただし,調節性分泌経路においては,テザリングの後にそのまま膜融合が起こると,内包される生理活性物質が非刺激時に分泌されてしまうため,何らかの機序で膜融合が抑制されていると考えられる.Granuphilinが結合するSyntaxin 1は,開放型と閉鎖型の構造をとることが知られており,C末端側のコイルドコイル領域をN末端側領域が覆い隠すような閉鎖型構造をとると,SNAP-25, Synaptobrevin 2など,他のSNARE分子と結合できなくなり,膜融合反応を引き起こせない.逆にN末端側領域とC末端側領域の間のヒンジと呼ばれる部分にあるロイシンとグルタミン酸をアラニンに変えると,Syntaxin 1は常にN末端とC末端が乖離した開放型構造をとる24).Granuphilinは,この常時開放型Syntaxin 1変異体と結合できないことから20),閉鎖型Syntaxin 1と特異的に結合して,膜融合反応を抑制すると考えられる.Granuphilinは,Munc18-1という分子とも結合するが25),Munc18-1は,Syntaxin 1と結合して,その閉鎖型構造を安定化する26).Munc18-1欠損副腎髄質細胞では,通常の電子顕微鏡標本で,明確な分泌顆粒の細胞膜ドッキング障害が認められる15).これらの知見は,Granuphilinが,分泌顆粒膜上のRab27と細胞膜上の閉鎖型Syntaxin 1とMunc18-1からなる複合体を橋渡しすることによって,分泌顆粒を細胞膜へドッキングさせることを示唆している(図1).しかし,Rab27を欠損する膵β細胞では,Granuphilin欠損細胞でみられるような,明確なドッキング障害が認められず,グルコース応答性インスリン分泌は逆に低下する27, 28).Rab27欠損細胞では,Rab27と近縁のRab3が,Granuphilinや,後述のGranuphilin非依存性ドッキング装置(図4および8節参照)と結合してドッキングをつかさどる可能性がある.また,Syntaxin 1欠損細胞の化学固定標本で,分泌顆粒のドッキング障害が必ずしも認められないのは,GranuphilinがSyntaxin 2やSyntaxin 3にも結合でき,これらのアイソフォームを介するドッキング作用が残存するためと考えられる11).閉鎖型Syntaxin/Munc18-1複合体は神経シナプス小胞のドッキングにも関与するとされているが29),シナプス小胞膜にはGranuphilinは発現しておらず,代わりにRab3のエフェクターRIM30)が,シナプス小胞と細胞膜の連結を担っていると考えられている(図2).Munc18-1など,SMと総称されるタンパク質は,すべての細胞内膜輸送経路の膜融合反応に必須の分子で,SNARE複合体形成のための鋳型またはシャペロンとして機能する31, 32).調節性分泌経路のドッキングに関わる閉鎖型Syntaxin/Munc18-1複合体が,どのようにその構造と結合様式を変化させて膜融合を引き起こすのかについては,後述するプライミングと関連し,精力的に研究されている33).
上記,Granuphilin欠損内分泌細胞の知見は,電子顕微鏡写真上の分泌顆粒の細胞内分布と,細胞集団から分泌されるホルモンの定量という,静的な計測に基づいている.開口放出反応におけるドッキングの機能的意義を調べるためには,生きた細胞でGranuphilinの動態とホルモンの開口放出を可視化して,直接,リアルタイムで観察することが望まれた.神経シナプス小胞の直径50 nmに比し,分泌顆粒の直径は300~350 nmくらいと大きいため,蛍光標識すれば,光学顕微鏡でも開口放出を観測することは可能である.しかし,細胞内には多数の分泌顆粒があるため,細胞膜で起こる開口放出現象を特異的に観察するためには,細胞内深部の顆粒からの蛍光を除去する必要があった.Almersらは,アクリジンオレンジによる酸性オルガネラの染色や,顆粒内のChromogranin BやNeuropeptide YとGFPを融合したタンパク質の発現により,分泌顆粒を標識し,エヴァネッセント場(カバーガラスすなわち細胞膜接着面から100~200 nm以内)の蛍光のみ捕らえる全反射蛍光顕微鏡を用いて,細胞膜近傍の顆粒から開口放出を観測した34, 35).また,Simonsらは,水疱性口内炎ウイルス由来の糖タンパク質VSVGの温度感受性変異体をYFPと融合して発現させ,構成性分泌経路における小胞膜が細胞膜に融合するようすを全反射顕微鏡で観察した36).それによると,多くの小胞は融合前に細胞膜近傍に短時間滞っているが,一部の小胞は,全反射顕微鏡で励起されない細胞膜より離れた部位から,飛び込んでくるかのように急速に細胞膜近傍に近づき,そのまま膜融合を起こすことを見いだした.次いで永松らは,膵β細胞株MIN6や単離マウス膵β細胞にインスリン-GFPを発現し,グルコース刺激後1~2分以内の比較的早期(第一相に相当)に起こる開口放出は,刺激前より細胞膜近傍にあった顆粒からのもので,それ以降,5~6分後(第二相に相当)に起こる開口放出は,細胞内深部から急速に細胞膜に近づき,そのまま膜融合を起こす顆粒からのものが多いことを示した37, 38).開口放出前のインスリン顆粒の挙動が必ずしも画一的ではないことは,筆者らを含む他の研究者によっても観察され28, 39),直線的な逐次連続モデルに必ずしも適合しない事象と考えられた.
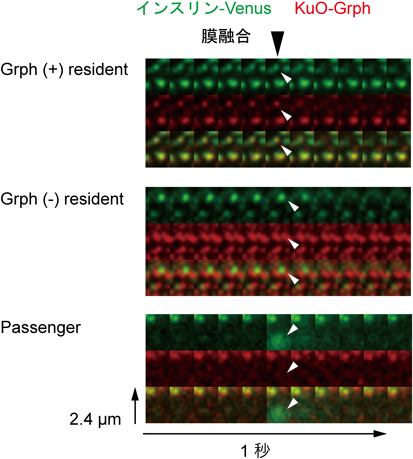
これらの知見をふまえ,筆者らは,Granuphilin欠損マウスより単離した膵β細胞にインスリン-GFPを発現させ,全反射顕微鏡で分泌顆粒の開口放出を56ミリ秒間隔で観察した(以下,文献28)の知見).電子顕微鏡写真で定義されるドッキング顆粒がほぼ消失しているGranuphilin欠損膵β細胞においても,野生型細胞より少ないものの,細胞膜近傍の顆粒が全反射顕微鏡で可視化された.刺激後の開口放出を,刺激前より細胞膜近傍にある顆粒(residentと呼称)と,エヴァネッセント場外から急速に動員される顆粒(passengerと呼称)に分別して観測すると(図3説明文参照),Granuphilin欠損細胞では,両タイプとも,刺激後早期から増大していることが判明した.また,野生型細胞ではほとんどみられない,静止期の開口放出の散発が,有意に増加していた.これらの知見より,全反射顕微鏡であらかじめ可視化されていた顆粒からの開口放出は,必ずしも細胞膜に接着したドッキング顆粒からのものとは限らないこと,分泌顆粒が分子的に細胞膜にドッキングされることは,刺激後早期の開口放出にも不要であること(もちろん膜融合時に,小胞膜と細胞膜の瞬間的な接着が必要であることは言うまでもない),さらに,Granuphilinによるドッキングが,細胞膜近傍の分泌顆粒が自発的に細胞膜と融合することを防いでいることを示している.
5. Granuphilinで細胞膜に繋留された顆粒からの開口放出
Granuphilinの知見が,ドッキングが膜融合を促進させるという逐次連続モデルに適合しないことから,開口放出可能で機能的なfunctional dockingとは別に,開口放出不能な行き止まりのdead-end dockingの存在が提唱された40).しかし筆者は,Granuphilin欠損細胞で細胞膜に接着した顆粒がほぼすべて消失することから,機能的ドッキングが別に存在する可能性は低く,Granuphilinによるドッキングを,刺激到来時まで自発的な開口放出を一時的に抑制する,生理的なものと考えた28, 41, 42).前述のように,全反射顕微鏡で可視化された顆粒は必ずしも細胞膜に接着(ドッキング)した顆粒とは限らないため,この問題を解決するには,分子的にGranuphilinによって細胞膜にテザリングされた顆粒と,単に細胞膜近傍にいる顆粒を区別して開口放出を計測する必要があった.そこで,インスリンをVenus, GranuphilinをKusabira-Orange 1(KuO)と,別々の蛍光タンパク質で標識したMIN6細胞で,Granuphilin陽性顆粒と陰性顆粒の開口放出を,全反射顕微鏡下,比較することを試みた.しかし内因性Granuphilinに加えてKuO-Granuphilinを発現すると,その過剰発現による抑制作用のため,開口放出現象の観察が困難となってしまった.そこで,MIN6など野生型膵β細胞株の作製法43, 44)に倣い,インスリン遺伝子のプロモーター下にSV40ウイルスlarge-T抗原を発現したトランスジェニックマウスと,Granuphilin欠損マウスを交配して,仔マウスより発生する膵島腫瘍(インスリノーマ)から,内因性Granuphilinを欠く膵β細胞株を樹立した.この膵β細胞株は,(Granuphilin欠損を反映して)野生型膵β細胞株に比べ,刺激後,2倍程度のインスリン分泌量を示したが,内因性レベルのKuO-Granuphilinを発現させると,野生型細胞株と同等の分泌量を示した(以下,文献45)の知見).この細胞を全反射顕微鏡で観察すると,インスリン-Venusで可視化された細胞膜近傍の顆粒の85%はGranuphilin陽性,15%は陰性で,内因性Granuphilinとインスリンを二重免疫染色したMIN6細胞を観察した際の陽性,陰性顆粒の比率とほぼ同等であった.静止期の陽性顆粒のxy平面における動きは,陰性顆粒のそれに比べてきわめて抑制されており,Granuphilinによって物理的に細胞膜につなぎ止められていることが示唆された.脱分極刺激を加えると,開口放出顆粒の16%は,エヴァネッセント場外から飛び込んで瞬時に開口放出するpassengerタイプ(図3下段)のものであった.このタイプの顆粒は,刺激前,細胞膜より離れた位置にいて,Granuphilinで細胞膜にテザリングされていないと考えられるが,開口放出時の1フレーム(2波長計測のため,103 ms)でしか観測できないため,その前にGranuphilinが局在していたかどうかを厳密には判定できない.そこで刺激前から可視化されていた細胞膜近傍の顆粒からのresidentタイプの開口放出84%に限定すると,40%はGranuphilin陽性顆粒(図3上段),44%は陰性顆粒(図3中段)であった.つまり,Granuphilinでテザリングされている陽性顆粒は,陰性顆粒とほぼ同数の開口放出を示し,決して,行き止まりの開口放出不能なものではなく,刺激に反応しうる機能的にドッキングした顆粒であることを示している.しかし,刺激前,可視化された細胞膜近傍にある顆粒の85%が陽性顆粒,15%が陰性顆粒であったことを考えると,陽性顆粒の開口放出効率(顆粒数あたりの開口放出数)は,陰性顆粒のそれに比べて20%以下(40/85÷44/15≒16)しかないことがわかる.また,陽性顆粒は,開口放出直前に,xy方向の動きが増え,Granuphilinの蛍光強度が減弱することから,Granuphilinが解離して,そのために可動性(動きの自由度)が大きくなることが示唆された.
ドッキング小胞を開口放出可能にするとされるプライミングは,歴史的には,二つの異なる研究手法により定義された.一つは,薬剤処理で細胞膜を透過性にした細胞に,刺激を加えて分泌反応がそれ以上起きなくなるようにした上で,外来性に細胞質抽出液を加え,Mg2+-ATP, Ca2+依存性の分泌を再び可能にさせる過程をプライミングと呼び,これに必要な成分を生化学的に同定する研究が行われた.その結果,細胞膜でのホスファチジルイノシトール二リン酸PIP2生成や46, 47),CAPS(線虫におけるUNC-31)という分子48, 49)が必要であることが判明した.しかし,このプライミングの定義は,必ずしも分泌小胞膜側の特性の変化を意味しておらず,細胞膜側や細胞質の必要条件を規定している可能性がある.もう一つの定義は,パッチクランプによる膜容量測定など,電気生理学手法によって,短時間の刺激により開口放出可能な小胞プール(readily releasable pool)を再充填する過程をプライミングと呼び,Munc13-1(線虫におけるUNC-13)50–52)などの分子が見いだされた.しかし,この定義の元となるreadily releasable poolは,論文によって異なる刺激や方法で規定されている.いずれにしても,これら二つのプライミングは,実験手法,分泌増加を測定するタイムスケール(分vsミリ秒~秒)がまったく異なり,同一の現象を対象にしているとは限らない.また,いずれの定義においても,ドッキングとの関係は定かではなく,逐次連続モデルで述べられている,プライミングが,ドッキングの後に起こり,刺激後のCa2+上昇前に起こっていることを保証していない.
前述のように,SNARE複合体形成による膜融合反応が起こるためには,Syntaxinの閉鎖型から開放型への移行が必須で,このことが,プライミング反応の実体(の一つ)である可能性がある.線虫のunc-13変異体でみられる神経シナプス伝達障害による麻痺が,野生型Syntaxinの過剰発現ではレスキューされないが,前述した常時開放型をとるSyntaxin変異体の発現によりレスキューされることから53),この過程にUNC-13が関わっていると考えられた.実際,Munc13-1のMUN領域が,in vitroで,閉鎖型Syntaxin 1/Munc18-1複合体を,開放型Syntaxin 1/SNAP-25/Synaptobrevin 2からなるSNARE複合体に変換させる活性があることが示されている54, 55).また,線虫のunc-10変異体(UNC-10哺乳類ホモログはRIM)では,膜融合可能な神経シナプス小胞の数は1/5に減少し,その伝達障害は,常時開放型Syntaxin変異体の発現によりレスキューされることが報告されている56).この知見は,RIMが,Munc13-1同様,プライミングに関わっていることを示唆している.実際,RIMは,ホモ二量体を形成している不活性型Munc13-1に結合して,これを単量体にして活性化すること,また,常時単量体をとるMunc13-1変異体は,RIM欠損シナプスの障害をレスキューすることが示され57),RIMがMunc13-1の上流で機能することが判明した(図2).
Munc13-1を欠損するマウスの神経細胞では,シナプス伝達が完全に喪失する50).一方,Munc13-1欠損膵β細胞では,持続的刺激による分泌は低下するが,刺激直後の分泌反応は正常で,分泌顆粒の分泌障害は部分的で,程度が軽い58).Munc13-1には,Ca2+やリン脂質と結合するC2領域や,プライミングに関与するMUN領域があるが,これらの領域を共通に持つ分子として,CAPSがあげられる.CAPSは,前節で述べたように,細胞膜を透過性にした内分泌細胞で分泌を促進する,第一の定義によるプライミング因子として見いだされたが,その後,CAPS欠損マウス神経細胞で,電気生理学的手法を用いた第二の定義による,シナプス小胞のプライミングにも関わっていることが報告された59).しかし,ショウジョウバエのCAPS欠損体のシナプス伝達障害は,分泌顆粒の開口放出障害の場合と異なり,細胞に野生型CAPSを発現させてもレスキューされないことから,神経細胞外の機能異常を反映する二次的なものと報告された60).また,線虫における変異体解析では,UNC-13が,シナプス小胞の開口放出に必須で,分泌顆粒の開口放出には不要であるのに対して52),UNC-31(CAPSホモログ)は,逆に分泌顆粒の開口放出にのみ関与すると報告された61, 62).これらの知見は,CAPSが,少なくとも進化的には,分泌顆粒の開口放出に第一義的に機能するとことを示唆している.また,CAPS欠失による分泌顆粒の開口放出障害の少なくとも一部は,常時開放型Syntaxinでレスキューされるが,Munc13-1ではレスキューされないことから,CAPSはSyntaxinの構造変化の過程に関わっているものの,Munc13-1の下流か,独立に機能していることが示唆された62, 63).一方,CAPS2欠損マウスの膵β細胞は,刺激後期のインスリン分泌障害を示すが,CAPS2より高発現しているCAPS1が残存するせいか,その程度は軽度であった64).前述のように,Granuphilinは,閉鎖型Syntaxinに結合して,ドッキング顆粒の膜融合反応を抑制すると考えられるため,Syntaxinを閉鎖型から開放型にすれば,ドッキング顆粒からGranuphilinが解離し,開口放出可能となりうる.しかし,この過程にMunc13-1やCAPSが関わっているかはわかっておらず,Granuphilinによる開口放出抑制を解除する機構の解明が待たれる.
8. Granuphilin非依存性の分泌顆粒ドッキング機構
前述のように,筆者は,Granuphilin欠損細胞の所見から,本分子が分泌顆粒の細胞膜ドッキングに必須と考え,別の機構の存在を想定していなかったが,最近,Rab27エフェクターの一つExophilin-8(別名MyRIP)がこの過程に関与する可能性が出てきた.Exophilin-8は,アクチン束上で機能するモータータンパク質,ミオシンVaやミオシンVIIaに結合活性を持ち,網膜色素細胞のメラノソームや内分泌細胞の分泌顆粒を,細胞辺縁部の皮質アクチン網に捕捉する役割を有することが示唆されていた65–69).皮質アクチン網は,トランスゴルジ網で生成された分泌顆粒が細胞膜と融合する際の障壁になりうるが,一方,分泌顆粒を細胞膜の近傍に保持・集積させることを可能にする.分泌小胞を積み荷とするアクチン上のモータータンパク質として,網膜メラノソームではミオシンVIIa70),分泌顆粒ではミオシンVa71–74)が,それぞれ機能することが示唆されていた.しかし,内分泌細胞内でExophilin-8とミオシンVaが実際に複合体を形成するという証明はなされておらず,MIN6細胞では,生理的条件下で内因性複合体は認められないと報告された75).
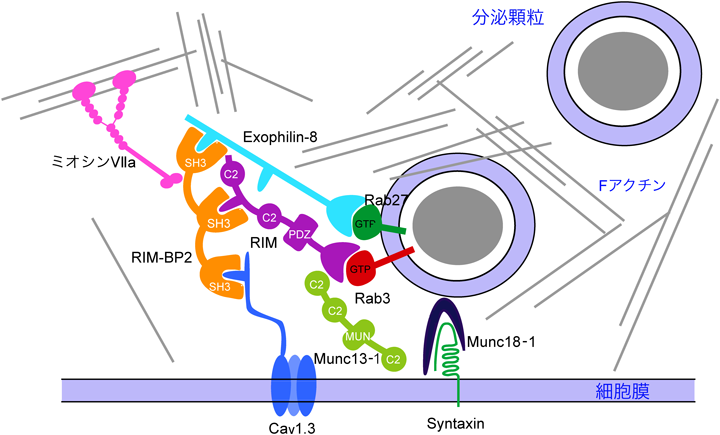
最近,筆者らは,膵β細胞で,Exophilin-8が,RIM-BP2と結合することを見いだした(以下,文献76)の所見).RIM-BP2は,もともとRIM結合分子として見いだされ77),RIMとともに,神経シナプス小胞と電位依存性Ca2+チャネルを橋渡しして,シナプス小胞を刺激後に生じるCa2+濃度の高いマイクロドメインに局在させ,迅速な開口放出を可能にすると考えられているが(図2)78, 79),内分泌細胞における機能はまったく不明であった.筆者らは,Exophilin-8が,ミオシンVaと直接結合するのではなく,RIM-BP2を介してミオシンVIIaと間接的に結合することによって,分泌顆粒を皮質アクチン網に捕捉・集積させることを示した.また,RIM-BP2は,神経細胞同様,膵β細胞において,RIMやL型Ca2+チャネルと結合していた.したがって,Exophilin-8−RIM-BP2−ミオシンVIIa複合体は,分泌顆粒を,単に物理的に皮質アクチン網に局在させるだけではなく,その開口放出に関与するRIMやL型Ca2+チャネル80, 81)との相互作用を仲介する働きがあると考えられた(図4).実際,Exophilin-8欠損マウス膵β細胞のインスリン分泌能は,野生型細胞に比し半減しており,また,培養膵β細胞株においても,本複合体形成による顆粒の細胞辺縁部集積作用は,刺激依存性ホルモン分泌量ときわめてよい相関を示した.RIM, RIM-BP2は,小胞膜上のRab3やExophilin-8,細胞膜上のCa2+チャネルや(Munc13-1を介して)Syntaxin 1と結合することから,分泌顆粒や神経シナプス小胞のドッキングに関与する可能性がある(図2, 4).事実,RIM欠損神経細胞の電子顕微鏡化学固定標本で,ドッキング小胞の減少が報告されている79, 82).また,Exophilin-8欠損マウス膵β細胞の化学固定標本においても,細胞膜直下の分泌顆粒の数は減少していたが,Granuphilin欠損細胞と比較すると,その減少の度合は小さく,細胞膜に接着した顆粒も残存していた.逐次連続モデルでは,ドッキング顆粒の一部のみ,何らかのプライミング反応を受けて開口放出可能となると想定されているが,ドッキングに関わるRab27エフェクターの種類により,細胞膜近傍の顆粒の開口放出効率が異なるという新たな可能性が出てきた.Exophilin-8によるRIMを介するドッキングは,シナプス小胞のドッキング機構と類似し,開口放出を促進させると考えられるが(図2, 4),今後,開口放出を抑制するGranuphilinによるドッキング機構(図1)との関係を明らかにする必要がある.
神経シナプス小胞は,Ca2+流入後100マイクロ秒以内に膜融合を起こすという報告もあり83),このような短時間に,小胞が細胞膜直下に移動してドッキングし,Syntaxinが閉鎖型から開放型になるプライミングが起こり,その後,SNARE複合体が形成されるとは考えにくい.神経シナプス小胞の即時応答性を考えると,刺激後に起こる事象は,Ca2+感知による膜融合反応のみと考えられる.本稿では詳述しないが,このCa2+感知には,Synaptotagminというタンパク質が関与し,最終的な膜融合反応を引き起こすと考えられている29).すなわち,刺激に即座に反応しうるシナプス小胞は,ドッキングやプライミングを終え,すでにSNARE複合体が形成されていると予想されることから(図5),小胞膜を細胞膜にデザリングさせる従来のドッキングと,プライミング反応を済ませ,SNARE複合体形成によって両膜を対向させる狭義のドッキングを,区別する必要があると考えられた.最近,高圧凍結法にトモグラフィーを組み合わせ,シナプス小胞膜と細胞膜間の距離が5 nm以内の小胞をテザリング小胞,同2 nm以内をドッキング小胞とする,精密な電子顕微鏡解析が行われた84).それによると,SNARE分子のSyntaxin 1, SNAP-25, Synaptobrevin-2,プライミングに機能するMunc13-1/2, CAPS1/2,それぞれを欠損する神経細胞において,2 nm以内の小胞の消失が認められることから,この狭義のドッキング小胞は,プライミングを終えて,膜融合を準備した状態と考えられた.これらの欠損細胞では,(その閉鎖型がデザリングにも関与する)Syntaxin 1を欠損する細胞を除いて,膜融合の抑制を反映して,細胞膜から5~40 nm離れた小胞は逆に増加していた.このため,0~40 nmの小胞の総数は変化せず,通常の電子顕微鏡解析でドッキング(テザリング)障害が認められない原因とされた.前述のように,RIM欠損神経細胞では,化学固定標本でシナプス小胞のドッキング障害が認められるが79, 82),RIMはプライミングにも関与することから56, 57),凍結トモグラフィー標本で狭義のドッキング障害も認められている85).このように高速の分泌反応を必要とする神経シナプス小胞においては,ドッキングとプライミングの両方に関与する分子があり,両者の境界があいまいになっている.
10. 調節性分泌経路における逐次連続モデルと並行モデル
最近,神経シナプスのアクティヴ・ゾーンでは,非刺激時においても小胞膜と細胞膜間にSNARE複合体が形成されているが,膵β細胞では,静止期には形成されておらず,刺激後に初めて形成されることが示された86).すなわち両細胞において,分泌小胞が刺激を待機している過程は異なっており,分泌顆粒は,静止期に狭義のドッキング(プライミング)の状態で待機していないことを示している.シナプス小胞開口放出のトリガーとなるSynaptotagminがCa2+を感知するC2ドメインは,Rab27エフェクター,RIM, Munc13, CAPSにもあり,遅い開口放出反応を示す分泌顆粒では,刺激直後と持続性刺激後のCa2+上昇によって誘導される律速段階が異なる可能性がある.また,ドッキング(デザリング)に関わるExophilin-8, Granuphilin,プライミングに関わるRIM-Munc13, CAPSなどの分子が,並行して機能することも考えられる.
全反射顕微鏡による開口放出前の分泌顆粒の動態観測によると,すべての顆粒が同一のルートで膜融合に至るとは限らないことが示唆される.これまで述べてきたように,分泌顆粒においては,通常の化学固定標本で見いだされる従来の定義によるドッキングは,開口放出に必須ではない.驚くべきことに,神経シナプス小胞においても,RIM, ELKS(アクティヴ・ゾーンの電子密度の高いところに存在するCAZと総称される分子の一つ)を同時に欠損させ,従来の定義によるドッキング小胞をほぼ完全に消失させても,開口放出可能なプールは残存するという87).筆者らがpassengerと呼称する,細胞内深部から飛び込んでくるようにみえる非ドッキング顆粒の開口放出は,開口放出した顆粒のオメガ構造が何らかの機序で安定的に残存しているところに膜融合を起こす,sequential exocytosisと呼ばれるもの88)も含まれていると思われるが,膵β細胞での2光子顕微鏡による観察では,その頻度は数%以下で89),passengerタイプの開口放出頻度よりはるかに低い.アクチン上のミオシンVaやVIIa,あるいは微小管上のキネシンが,顆粒を細胞内深部から細胞骨格上で運搬し,この過程にExophilin-790, 91),Exophilin-8,さらに別のRab27エフェクターが関与する可能性もある.しかし,同一細胞に発現する複数のエフェクターが,同じRab27という分子と結合しながら,どのように異なるステップを制御しているのか,不明な点が多い.
これまで述べてきた知見より,筆者は,少なくとも分泌顆粒の開口放出過程においては,直線的な逐次連続モデル(linear/sequential model)が必ずしも当てはまらず,ドッキング顆粒と非ドッキング顆粒が,複数のルートと分子機構により,並行して開口放出するモデル(parallel model)を提唱している41, 42).高速の神経シナプス伝達の研究が先行していることもあり,依然,画一的な逐次連続モデルが調節性分泌経路に共通する機構として信じてられているが40),分泌顆粒独自の開口放出過程に関する知見を積み重ねていく必要がある.
謝辞Acknowledgments
本稿で紹介した筆者らの研究は,群馬大学生体調節研究所遺伝生化学分野において,多くの同僚,共同研究者の方々とともに行われたものです.また,本稿の図作成に関しては,助教の水野広一さんと,秘書の新後閑幸子さんに協力していただきました.この場を借りて深く感謝申し上げます.
引用文献References
1) Kasai, H. (1999) Trends Neurosci., 22, 88–93.
2) Izumi, T., Gomi, H., Kasai, K., Mizutani, S., & Torii, S. (2003) Cell Struct. Funct., 28, 465–474.
3) Izumi, T. (2007) Endocr. J., 54, 649–657.
4) Broadie, K., Prokop, A., Bellen, H.J., O’Kane, C.J., Schulze, K.L., & Sweeney, S.T. (1995) Neuron, 15, 663–673.
5) Borisovska, M., Zhao, Y., Tsytsyura, Y., Glyvuk, N., Takamori, S., Matti, U., Rettig, J., Südhof, T., & Bruns, D. (2005) EMBO J., 24, 2114–2126.
6) Sørensen, J.B., Nagy, G., Varoqueaux, F., Nehring, R.B., Brose, N., Wilson, M.C., & Neher, E. (2003) Cell, 114, 75–86.
7) O’Connor, V., Heuss, C., De Bello, W.M., Dresbach, T., Charlton, M.P., Hunt, J.H., Pellegrini, L.L., Hodel, A., Burger, M.M., Betz, H., Augustine, G.J., & Schäfer, T. (1997) Proc. Natl. Acad. Sci. USA, 94, 12186–12191.
8) Marsal, J., Ruiz-Montasell, B., Blasi, J., Moreira, J.E., Contreras, D., Sugimori, M., & Llinás, R. (1997) Proc. Natl. Acad. Sci. USA, 94, 14871–14876.
9) de Wit, H., Cornelisse, L.N., Toonen, R.F., & Verhage, M. (2006) PLoS One, 1, e126.
10) Ohara-Imaizumi, M., Fujiwara, T., Nakamichi, Y., Okamura, T., Akimoto, Y., Kawai, J., Matsushima, S., Kawakami, H., Watanabe, T., Akagawa, K., & Nagamatsu, S. (2007) J. Cell Biol., 177, 695–705.
11) Wang, H., Ishizaki, R., Kobayashi, E., Fujiwara, T., Akagawa, K., & Izumi, T. (2011) J. Biol. Chem., 286, 32244–32250.
12) de Wit, H., Walter, A.M., Milosevic, I., Gulyás-Kovács, A., Riedel, D., Sørensen, J.B., & Verhage, M. (2009) Cell, 138, 935–946.
13) Hammarlund, M., Palfreyman, M.T., Watanabe, S., Olsen, S., & Jorgensen, E.M. (2007) PLoS Biol., 5, e198.
14) Wu, Y., Gu, Y., Morphew, M.K., Yao, J., Yeh, F.L., Dong, M., & Chapman, E.R. (2012) J. Cell Biol., 198, 323–330.
15) Voets, T., Toonen, R.F., Brian, E.C., de Wit, H., Moser, T., Rettig, J., Südhof, T.C., Neher, E., & Verhage, M. (2001) Neuron, 31, 581–591.
16) Gomi, H., Mizutani, S., Kasai, K., Itohara, S., & Izumi, T. (2005) J. Cell Biol., 171, 99–109.
17) Wang, J., Takeuchi, T., Yokota, H., & Izumi, T. (1999) J. Biol. Chem., 274, 28542–28548.
18) Yi, Z., Yokota, H., Torii, S., Aoki, T., Hosaka, M., Zhao, S., Takata, K., Takeuchi, T., & Izumi, T. (2002) Mol. Cell. Biol., 22, 1858–1867.
19) Zhao, S., Torii, S., Yokota-Hashimoto, H., Takeuchi, T., & Izumi, T. (2002) Endocrinology, 143, 1817–1824.
20) Torii, S., Zhao, S., Yi, Z., Takeuchi, T., & Izumi, T. (2002) Mol. Cell. Biol., 22, 5518–5526.
21) Torii, S., Takeuchi, T., Nagamatsu, S., & Izumi, T. (2004) J. Biol. Chem., 279, 22532–22538.
22) Gomi, H., Mori, K., Itohara, S., & Izumi, T. (2007) Mol. Biol. Cell, 18, 4377–4386.
23) Grosshans, B.L., Ortiz, D., & Novick, P. (2006) Proc. Natl. Acad. Sci. USA, 103, 11821–11827.
24) Dulubova, I., Sugita, S., Hill, S., Hosaka, M., Fernandez, I., Südhof, T.C., & Rizo, J. (1999) EMBO J., 18, 4372–4382.
25) Coppola, T., Frantz, C., Perret-Menoud, V., Gattesco, S., Hirling, H., & Regazzi, R. (2002) Mol. Biol. Cell, 13, 1906–1915.
26) Misura, K.M., Scheller, R.H., & Weis, W.I. (2000) Nature, 404, 355–362.
27) Kasai, K., Ohara-Imaizumi, M., Takahashi, N., Mizutani, S., Zhao, S., Kikuta, T., Kasai, H., Nagamatsu, S., Gomi, H., & Izumi, T. (2005) J. Clin. Invest., 115, 388–396.
28) Kasai, K., Fujita, T., Gomi, H., & Izumi, T. (2008) Traffic, 9, 1191–1203.
29) Südhof, T.C. (2013) Neuron, 80, 675–690.
30) Wang, Y., Okamoto, M., Schmitz, F., Hofmann, K., & Südhof, T.C. (1997) Nature, 388, 593–598.
31) Baker, R.W., Jeffrey, P.D., Zick, M., Phillips, B.P., Wickner, W.T., & Hughson, F.M. (2015) Science, 349, 1111–1114.
32) Ma, L., Rebane, A.A., Yang, G., Xi, Z., Kang, Y., Gao, Y., & Zhang, Y. (2015) eLife, 4, e09580.
33) Rizo, J. & Südhof, T.C. (2012) Annu. Rev. Cell Dev. Biol., 28, 279–308.
34) Steyer, J.A., Horstmann, H., & Almers, W. (1997) Nature, 388, 474–478.
35) Lang, T., Wacker, I., Steyer, J., Kaether, C., Wunderlich, I., Soldati, T., Gerdes, H.H., & Almers, W. (1997) Neuron, 18, 857–863.
36) Toomre, D., Steyer, J.A., Keller, P., Almers, W., & Simons, K. (2000) J. Cell Biol., 149, 33–40.
37) Ohara-Imaizumi, M., Nakamichi, Y., Tanaka, T., Ishida, H., & Nagamatsu, S. (2002) J. Biol. Chem., 277, 3805–3808.
38) Ohara-Imaizumi, M., Nishiwaki, C., Kikuta, T., Nagai, S., Nakamichi, Y., & Nagamatsu, S. (2004) Biochem. J., 381, 13–18.
39) Shibasaki, T., Takahashi, H., Miki, T., Sunaga, Y., Matsumura, K., Yamanaka, M., Zhang, C., Tamamoto, A., Satoh, T., Miyazaki, J., & Seino, S. (2007) Proc. Natl. Acad. Sci. USA, 104, 19333–19338.
40) Verhage, M. & Sørensen, J.B. (2008) Traffic, 9, 1414–1424.
41) Izumi, T., Kasai, K., & Gomi, H. (2007) Diabetes Obes. Metab., 9(Suppl 2), 109–117.
42) Izumi, T. (2011) Front. Biosci. (Landmark Ed.), 16, 360–367.
43) Hanahan, D. (1985) Nature, 315, 115–122.
44) Miyazaki, J., Araki, K., Yamato, E., Ikegami, H., Asano, T., Shibasaki, Y., Oka, Y., & Yamamura, K. (1990) Endocrinology, 127, 126–132.
45) Mizuno, K., Fujita, T., Gomi, H., & Izumi, T. (2016) Sci. Rep., 6, 23909.
46) Eberhard, D.A., Cooper, C.L., Low, M.G., & Holz, R.W. (1990) Biochem. J., 268, 15–25.
47) Hay, J.C., Fisette, P.L., Jenkins, G.H., Fukami, K., Takenawa, T., Anderson, R.A., & Martin, T.F. (1995) Nature, 374, 173–177.
48) Walent, J.H., Porter, B.W., & Martin, T.F. (1992) Cell, 70, 765–775.
49) Ann, K., Kowalchyk, J.A., Loyet, K.M., & Martin, T.F. (1997) J. Biol. Chem., 272, 19637–19640.
50) Augustin, I., Rosenmund, C., Südhof, T.C., & Brose, N. (1999) Nature, 400, 457–461.
51) Aravamudan, B., Fergestad, T., Davis, W.S., Rodesch, C.K., & Broadie, K. (1999) Nat. Neurosci., 2, 965–971.
52) Richmond, J.E., Davis, W.S., & Jorgensen, E.M. (1999) Nat. Neurosci., 2, 959–964.
53) Richmond, J.E., Weimer, R.M., & Jorgensen, E.M. (2001) Nature, 412, 338–341.
54) Ma, C., Li, W., Xu, Y., & Rizo, J. (2011) Nat. Struct. Mol. Biol., 18, 542–549.
55) Ma, C., Su, L., Seven, A.B., Xu, Y., & Rizo, J. (2013) Science, 339, 421–425.
56) Koushika, S.P., Richmond, J.E., Hadwiger, G., Weimer, R.M., Jorgensen, E.M., & Nonet, M.L. (2001) Nat. Neurosci., 4, 997–1005.
57) Deng, L., Kaeser, P.S., Xu, W., & Südhof, T.C. (2011) Neuron, 69, 317–331.
58) Kang, L., He, Z., Xu, P., Fan, J., Betz, A., Brose, N., & Xu, T. (2006) Cell Metab., 3, 463–468.
59) Jockusch, W.J., Speidel, D., Sigler, A., Sørensen, J.B., Varoqueaux, F., Rhee, J.S., & Brose, N. (2007) Cell, 131, 796–808.
60) Renden, R., Berwin, B., Davis, W., Ann, K., Chin, C.T., Kreber, R., Ganetzky, B., Martin, T.F., & Broadie, K. (2001) Neuron, 31, 421–437.
61) Speese, S., Petrie, M., Schuske, K., Ailion, M., Ann, K., Iwasaki, K., Jorgensen, E.M., & Martin, T.F. (2007) J. Neurosci., 27, 6150–6162.
62) Hammarlund, M., Watanabe, S., Schuske, K., & Jorgensen, E.M. (2008) J. Cell Biol., 180, 483–491.
63) Liu, Y., Schirra, C., Edelmann, L., Matti, U., Rhee, J., Hof, D., Bruns, D., Brose, N., Rieger, H., Stevens, D.R., & Rettig, J. (2010) J. Cell Biol., 190, 1067–1077.
64) Speidel, D., Salehi, A., Obermueller, S., Lundquist, I., Brose, N., Renström, E., & Rorsman, P. (2008) Cell Metab., 7, 57–67.
65) El-Amraoui, A., Schonn, J.S., Küssel-Andermann, P., Blanchard, S., Desnos, C., Henry, J.P., Wolfrum, U., Darchen, F., & Petit, C. (2002) EMBO Rep., 3, 463–470.
66) Desnos, C., Schonn, J.S., Huet, S., Tran, V.S., El-Amraoui, A., Raposo, G., Fanget, I., Chapuis, C., Ménasché, G., de Saint Basile, G., Petit, C., Cribier, S., Henry, J.P., & Darchen, F. (2003) J. Cell Biol., 163, 559–570.
67) Waselle, L., Coppola, T., Fukuda, M., Iezzi, M., El-Amraoui, A., Petit, C., & Regazzi, R. (2003) Mol. Biol. Cell, 14, 4103–4113.
68) Mizuno, K., Ramalho, J.S., & Izumi, T. (2011) Mol. Biol. Cell, 22, 1716–1726.
69) Huet, S., Fanget, I., Jouannot, O., Meireles, P., Zeiske, T., Larochette, N., Darchen, F., & Desnos, C. (2012) J. Neurosci., 32, 2564–2577.
70) Liu, X., Ondek, B., & Williams, D.S. (1998) Nat. Genet., 19, 117–118.
71) Rudolf, R., Kögel, T., Kuznetsov, S.A., Salm, T., Schlicker, O., Hellwig, A., Hammer, J.A. III, & Gerdes, H.H. (2003) J. Cell Sci., 116, 1339–1348.
72) Ivarsson, R., Jing, X., Waselle, L., Regazzi, R., & Renström, E. (2005) Traffic, 6, 1027–1035.
73) Varadi, A., Tsuboi, T., & Rutter, G.A. (2005) Mol. Biol. Cell, 16, 2670–2680.
74) Desnos, C., Huet, S., Fanget, I., Chapuis, C., Böttiger, C., Racine, V., Sibarita, J.B., Henry, J.P., & Darchen, F. (2007) J. Neurosci., 27, 10636–10645.
75) Brozzi, F., Lajus, S., Diraison, F., Rajatileka, S., Hayward, K., Regazzi, R., Molnár, E., & Váradi, A. (2012) Mol. Biol. Cell, 23, 4444–4455.
76) Fan, F., Matsunaga, K., Wang, H., Ishizaki, R., Kobayashi, E., Kiyonari, H., Mukumoto, Y., Okunishi, K., & Izumi, T. (2017) eLife, 6, e26174.
77) Wang, Y., Sugita, S., & Südhof, T.C. (2000) J. Biol. Chem., 275, 20033–20044.
78) Hibino, H., Pironkova, R., Onwumere, O., Vologodskaia, M., Hudspeth, A.J., & Lesage, F. (2002) Neuron, 34, 411–423.
79) Kaeser, P.S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., & Südhof, T.C. (2011) Cell, 144, 282–295.
80) Yang, S.N. & Berggren, P.O. (2006) Endocr. Rev., 27, 621–676.
81) Yasuda, T., Shibasaki, T., Minami, K., Takahashi, H., Mizoguchi, A., Uriu, Y., Numata, T., Mori, Y., Miyazaki, J., Miki, T., & Seino, S. (2010) Cell Metab., 12, 117–129.
82) Han, Y., Kaeser, P.S., Südhof, T.C., & Schneggenburger, R. (2011) Neuron, 69, 304–316.
83) Sabatini, B.L. & Regehr, W.G. (1996) Nature, 384, 170–172.
84) Imig, C., Min, S.W., Krinner, S., Arancillo, M., Rosenmund, C., Sudhof, T.C., Rhee, J., Brose, N., & Cooper, B.H. (2014) Neuron, 84, 416–431.
85) Fernández-Busnadiego, R., Asano, S., Oprisoreanu, A.M., Sakata, E., Doengi, M., Kochovski, Z., Zürner, M., Stein, V., Schoch, S., Baumeister, W., & Lučić, V. (2013) J. Cell Biol., 201, 725–740.
86) Takahashi, N., Sawada, W., Noguchi, J., Watanabe, S., Ucar, H., Hayashi-Takagi, A., Yagishita, S., Ohno, M., Tokumaru, H., & Kasai, H. (2015) Nat. Commun., 6, 8531.
87) Wang, S.S., Held, R.G., Wong, M.Y., Liu, C., Karakhanyan, A., & Kaeser, P.S. (2016) Neuron, 91, 777–791.
88) Pickett, J.A. & Edwardson, J.M. (2006) Traffic, 7, 109–116.
89) Takahashi, N., Hatakeyama, H., Okado, H., Miwa, A., Kishimoto, T., Kojima, T., Abe, T., & Kasai, H. (2004) J. Cell Biol., 165, 255–262.
90) Arimura, N., Kimura, T., Nakamuta, S., Taya, S., Funahashi, Y., Hattori, A., Shimada, A., Ménager, C., Kawabata, S., Fujii, K., Iwamatsu, A., Segal, R.A., Fukuda, M., & Kaibuchi, K. (2009) Dev. Cell, 16, 675–686.
91) Wang, H., Ishizaki, R., Xu, J., Kasai, K., Kobayashi, E., Gomi, H., & Izumi, T. (2013) Mol. Biol. Cell, 24, 319–330.
著者紹介Author Profile
 泉 哲郎(いずみ てつろう)
泉 哲郎(いずみ てつろう)群馬大学生体調節研究所教授(遺伝生化学分野).医学博士.
略歴1957年長崎県に生る.82年東京大学医学部医学科卒業.89年東京大学医学部第三内科助手.90年米国コロラド大学医学部HHMI留学.94年群馬大学生体調節研究所助教授.2000年より現職.
研究テーマと抱負糖尿病や肥満の成因・病態生理の一端解明につながることを願い,1)調節性分泌経路,特にインスリン分泌顆粒の開口放出,2)脂肪細胞における脂肪蓄積,の分子機序を,細胞生物学,マウス遺伝学の手法を用いて研究している.
ウェブサイトhttp://molend.showa.gunma-u.ac.jp/
趣味音楽鑑賞,ピアノ演奏.