ペルオキシソーム(peroxisome)は,一重の膜で囲まれた酵母からヒトまで真核細胞に広く存在する直径0.1~1.5 µmの細胞小器官(オルガネラ)である.この小器官は,1954年Rhodinによってマウス腎臓尿細管上皮細胞内に形態学的に見いだされた.1960年代初期までにラット肝臓ペルオキシソームに尿酸オキシダーゼ,D-アミノ酸オキシダーゼ,L-α-ヒドロキシ酸オキシダーゼなどの過酸化水素(H2O2)を生じるオキシダーゼ群とH2O2を分解するカタラーゼが局在することが明らかとなり,de Duveらは,この細胞小器官の機能的名称としてペルオキシソームと命名し1),今日この名称が広く定着している.
高等動物ペルオキシソームの機能研究は,Lazarowとde Duveがラット肝臓ペルオキシソームにおいて,ミトコンドリアに局在するものとは異なる脂肪酸のβ-酸化系(グリオキシソームのβ-酸化系と同じ)を発見したことにより大きく前進した2).一方,新生児期より脳・肝臓などの障害に加え筋緊張低下,顔貌異常,精神運動発達遅延など多発奇形を有するZellweger症候群の患者では,Goldfischerらによって形態学的にペルオキシソームが観察されないことが報告された.さらに,Moserらのグループによって,Zellweger症候群患者では,極長鎖脂肪酸が蓄積されること3, 4),次いでBorstらによりエーテルリン脂質プラスマローゲンの著しい減少5)が報告されるなど,ペルオキシソームに特有な脂質代謝の障害と病態の発症機構との関連が注目された.
ヒト先天性ペルオキシソーム欠損症の一次的病因の同定に関して著者らのグループは,チャイニーズハムスター卵巣(CHO)細胞からヒトペルオキシソーム欠損症患児由来細胞と同じ表現型を示すペルオキシソーム欠損性CHO変異細胞の分離,次いでペルオキシソームの形成回復を指標とした機能相補性スクリーニング法による原因遺伝子の単離法を確立した.加えて,その後確立されたペルオキシソーム欠損性酵母の相補遺伝子のヒトホモログ検索法(EST法)の相補的活用により,ヒト先天性ペルオキシソーム欠損症の10以上に及ぶ病因遺伝子を解明した.現在では,多くの研究グループによる成果も含めて14の相補性群に分類されるZellweger症候群のすべての原因遺伝子の同定が達成されている.また,それらの遺伝子産物であるペルオキシソーム形成因子(ペルオキシン,PEX因子)の機能解明も精力的になされている6, 7).一方,これら致死性ペルオキシソーム形成不全症に加え,ペルオキシソームのβ-酸化反応やプラスマローゲンの合成を触媒する酵素の単一遺伝子疾患などの病態およびそれらの原因遺伝子の同定も,並行して展開されてきた8, 9).これらの遺伝性疾患の代謝異常解明の研究成果から,高等動物のペルオキシソームは極長鎖脂肪酸,ジカルボン酸(DCA)のβ-酸化や分岐鎖脂肪酸のα-酸化などの異化作用,プラスマローゲン,胆汁酸やドコサヘキサエン酸(DHA, C22:6n-3)の合成などの同化作用を担うと理解されているが,ペルオキシームの脂肪酸β-酸化反応の生理的条件下での制御機構は不明なままである.
本稿では,高等動物におけるペルオキシソームの恒常性(生合成とペキソファジー)および生理機能とその制御機構に関して,我々のグループの成果を中心に概説する.
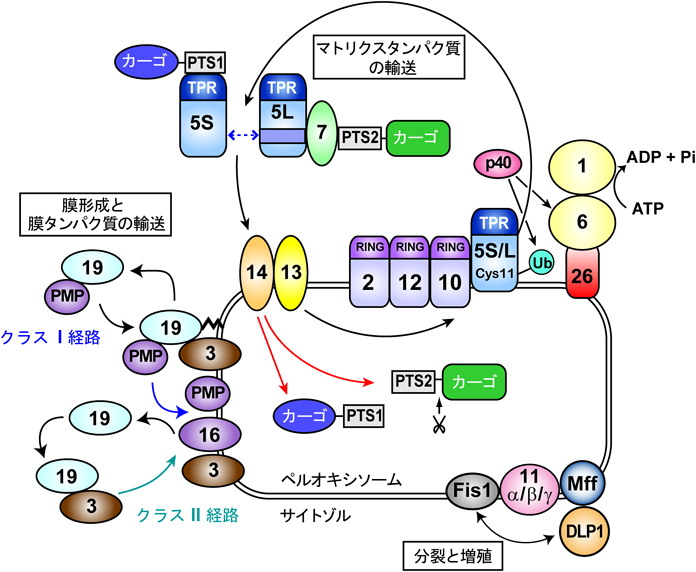
ペルオキシソームに局在するタンパク質はすべて核ゲノムにコードされており,サイトゾルの遊離型ポリソームで新規合成された構成タンパク質が既存のペルオキシソームに局在化し,その結果ペルオキシソームが成長,分裂して増殖していくという“growth and division model”がペルオキシソームの形成機構として一般的に受け入れられている10).また,近年ペルオキシソーム特異的オートファジー[ペキソファジー(pexophagy)]を介した分解機構を含め,これらの統合的制御によりペルオキシソームの恒常性が維持されていると考えられている.ペルオキシン群の機能解析も大きく進展し,ペルオキシンの機能は,1)マトリクスタンパク質輸送,2)膜タンパク質輸送と初期膜形成,3)分裂と形態制御,の三つに大別されることが明らかとなった(図1)11, 12).
ペルオキシソームマトリクスタンパク質の輸送シグナルとして,現在までにC末端アミノ酸配列-Ser-Lys-Leu(SKLモチーフ)を典型とする非切断型のPTS1(peroxisome-targeting signal 1),N末端に保存性の高い9アミノ酸を含有する切断型のPTS2が同定されており,大半の酵素はPTS1型である.PTS1タンパク質はPTS1受容体であるPex5により運ばれるが,PTS2タンパク質はPex7を介してPex5Sの内部に37アミノ酸の挿入配列を有するPex5LとPex5L-Pex7-PTS2カーゴの複合体として輸送され,Pex14/13で構成される膜透過装置を経てペルオキソーム内へ局在化される.PTSカーゴを遊離したPex5S/Lは,C末端にRINGフィンガーを有する3種のRINGペルオキシンPex2, Pex10, Pex12を含む複合体を経由し,N末端領域のシステイン残基にモノユビキチン化修飾13)を受け,AAAファミリータンパク質Pex1, Pex6およびATP加水分解活性依存的にサイトゾルにエクスポートされる.Awp1/ZFAND6(p40と略)はユビキチン化修飾型Pex5とPex6に結合し,Pex5のエクスポートを正の方向に制御していると考えられる14, 15).
ペルオキシソーム膜形成過程の必須因子はPex3, Pex16, Pex19である.Pex3を除くほとんどのペルオキソーム膜タンパク質はクラスI経路を介してPex19依存的にPex3へ標的化される16–18).一方,Pex3はクラスII経路によりPex19依存的にPex16へ標的化される19).C末端近傍に一つの膜貫通ドメインを有するテイルアンカー(TA)型膜タンパク質の哺乳動物ペルオキシソームへの標的化は,Pex26をはじめ複数のTAタンパク質をモデルとして詳細に解析され,中程度の疎水性膜貫通領域および正電荷を帯びたC末端側領域に依存した上記クラスI経路での標的化機構が明らかにされている17).一方,酵母の代表的なペルオキシソーム局在性TA膜タンパク質であるPex15(Pex26オーソログ)は,guided entry of tail-anchored proteins(GET)systemを介する小胞体への標的化を経由しペルオキシソームへと局在化するとの報告20, 21)は興味深い.
ペルオキシソームの分裂には,3種のPex11アイソフォームとミトコンドリアの分裂にも機能するダイナミン様タンパク質1(DLP1),MffおよびFis1が関わる.ペルオキシソーム分裂にはペルオキシソームの代謝産物であるDHAを含む脂質依存的なPex11βの複合体化が必要であるが,分裂制御機構の詳細は不明である22, 23).
哺乳類での選択的オートファジーでは,とくにユビキチン化を介した経路がよく研究されている.ペルオキシソームやミトコンドリアについてもユビキチンを介した選択的オートファジーによる分解が報告されてきた24, 25).ユビキチンを融合したペルオキシソーム膜タンパク質の発現24),ペルオキシソーム膜タンパク質Pex3の過剰発現による膜上タンパク質のユビキチン化を想定したペキソファジーが報告された26).さらに,ペルオキシソームマトリクスタンパク質の輸送を担うPex5のユビキチン化は,ペルオキシソーム生合成の制御だけでなく,ペキソファジーに関与することも報告されている27, 28).ペルオキシソームで産生された活性酸素種依存的なataxia telangiectasia mutated(ATM)キナーゼの活性化やアミノ酸飢餓条件においてPex5は,RINGペルオキシン複合体を形成するPex2, Pex10, Pex12のE3ユビキチンリガーゼ活性によってユビキチン化される.ペキソファジーに関する今後のより詳細な分子機構の解明が待たれる.
4. 高等動物におけるペルオキシソームの脂肪酸のβ-酸化反応
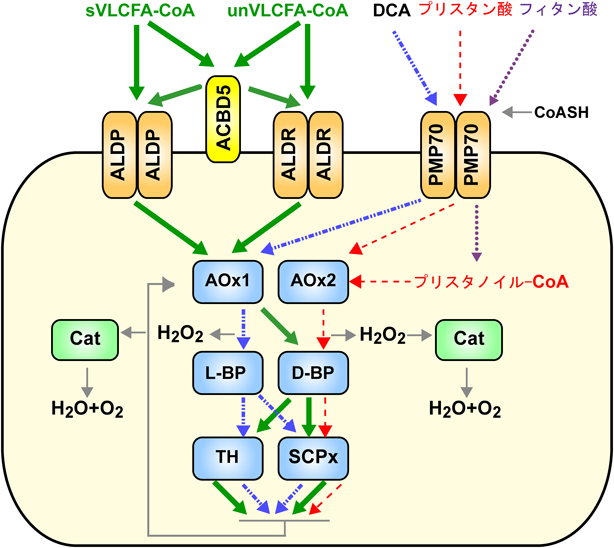
哺乳類においてアシル-CoAのカルボキシ端から順次2個ずつ炭素鎖が切り出される異化経路であるβ-酸化反応は,ミトコンドリアとペルオキシソームでなされる.炭素数22以上の極長鎖脂肪酸-CoAはペルオキシソームに特有なβ-酸化経路によって分解され,鎖長の短くなったアシル-CoAはミトコンドリアでアセチル-CoAにまで分解されて細胞のエネルギー源となる.いずれの細胞小器官のβ-酸化においても,アシル-CoAが脱水素反応,水付加反応,脱水素反応,チオール開裂反応によってアセチル-CoAと炭素数が二つ少ないアシル-CoAとなる(図2).しかしながら,ミトコンドリアとペルオキシソームにおけるβ-酸化反応はまったく異なる酵素によって触媒される.
1)ABCトランスポーターとアシル-CoA結合タンパク質
ペルオキシソームでは,飽和型および不飽和型の極長鎖脂肪酸,DCAのCoA誘導体がβ-酸化で分解される.一方,β位にメチル基が付加している代表的な分岐鎖脂肪酸であるフィタン酸,胆汁酸合成の中間産物である3α,7α-ジヒドロキシ-5β-コレスタン酸(DHCA)や3α,7α,12α-トリヒドロキシ-5β-コレスタン酸(THCA)のCoA誘導体は,ペルオキシソームにおいてα炭素が水酸化されたのちCO2として遊離するα-酸化反応を受け,続いてβ-酸化で分解される.これらペルオキシソームマトリクスで分解される基質は,基質特異性の異なる3種のペルオキシソーム局在性ABCトランスポーター(ATP-binding cassette transporter)であるALDP, ALDR, PMP70依存的にペルオキシソームマトリクスへと取り込まれる.多くのトランスポーターは12の膜貫通領域を有するのに対して,ペルオキシソームに局在するこれらのトランスポーターは六つの膜貫通領域を有しており,ホモ二量体を形成して機能を発揮すると考えられている29).ただALDPとALDR間でヘテロ二量体を形成するとも報告されている30).ごく最近,著者らを含む二つの研究グループは,網膜変性症の病因因子であるアシル-CoA結合ドメイン含有タンパク質5(acyl-CoA binding domain-containing 5:ACBD5)の遺伝子欠損細胞において飽和型だけでなく不和飽和型の極長鎖脂肪酸を有するホスファチジルコリンが増加することを見いだした31, 32).さらに,ACBD5はペルオキシソーム局在性TA膜タンパク質であることを見いだし,ACBD5のアシル-CoA結合ドメインを介してペルオキシソーム内への極長鎖アシル-CoAの取り込みとβ-酸化反応に重要な働きを持っていることを明らかにした31, 32).また,ペルオキシソーム膜には,ACBD5と58%の相同性を有し,ドメイン構造が類似するACBD4の存在も知られている33)ことから,これら2種のACBDは,異なるCoA誘導体のABCトランポーターへの輸送を担うものと推察される.
ペルオキシソーム膜トランスポーターであるALDPの機能不全によって極長鎖脂肪酸-CoAのペルオキシソームマトリクスへの輸送が障害されるため,極長鎖脂肪酸を有するリン脂質の蓄積などを特徴とする副腎白質ジストロフィー(X-linked adrenoleukodystrophy)を発症することが報告されている34).近年,PMP70の機能不全により分岐鎖脂肪酸,DHC-CoAやTHC-CoAのペルオキシソームへの取り込み障害の報告により,ペルオキシソーム膜の主要な構成タンパク質であるPMP70の機能が明らかとなった35).一方,ALDRの機能不全を原因とするヒト疾患については,まだ報告がない.
2)アシル-CoAオキシダーゼ
ペルオキシソームβ-酸化系サイクルの初発反応はアシル-CoAオキシダーゼ(acyl-CoA oxidase)によって触媒される.この反応では直接O2分子がH2O2の生成を伴い,アシル-CoAはエノイル-CoAとなる.ここで発生したH2O2はマトリクス酵素カタラーゼにより分解,あるいは亜硝酸塩などが存在する場合にはカタラーゼによる酸化反応に利用される.ラット肝臓より精製したアシル-CoAオキシダーゼ1(AOx1)はFAD酵素で,その分子量は150 kDaであり,一般的にA, B, Cと呼称される三つのポリペプチド鎖からなる.これらA, B, Cのポリペプチド鎖はそれぞれ75 kDa, 53 kDa, 22 kDaである.ペルオキシソーム形成不全症であるZellweger症候群患者由来の線維芽細胞では,サイトゾルにA鎖のみが検出される.また,放射性アミノ酸を用いたパルスチェイス実験ではまずA鎖が検出され,その後BおよびC鎖が検出される.これらのことから本酵素は,まずAポリペプチドとして合成され,ペルオキシソーム内に移行した後,Aポリペプチド分子の一部がプロテアーゼTysnd1(trypsin domain containing protein 1)36, 37)によるプロセシングを受けてBとCのポリペプチド鎖が生成,A2,ABC, B2C2のヘテロ二量体として機能する.一方,AOx2はα-酸化反応により分解された基質のβ-酸化反応を担う.本酵素はAOx1とは異なりペルオキシソームマトリクスにおける切断は受けない8).
3)二頭酵素
ペルオキシソームでは78 kDaの一本鎖のポリペプチドがβ-酸化反応の2番目のステップであるエノイル-CoAヒドラターゼ(enoyl-CoA hydratase)と3-ヒドロキシアシル-CoAデヒドロゲナーゼ(3-hydroxyacyl-CoA dehydrogenase)の二つの反応を触媒している.したがって,ペルオキシソームのこの酵素は正式にはエノイル-CoAヒドラターゼ-3-ヒドロキシアシル-CoAデヒドロゲナーゼ二頭酵素(enoyl-CoA hydratase-3-hydroxyacyl-CoA dehydrogenase bifunctional enzyme)という.一般には二頭酵素(bifunctional enzyme)と略称される.ペルオキシソームマトリクスには活性は同じであるが光学的性質の異なる中間体3-ヒドロキシアシル-CoAを産生し3-ケトアシル-CoAを産生する2種の二頭酵素L-BP(L-bifunctional protein)およびD-BP(D-bifunctional protein)が存在する.D-BPはペルオキシソームβ-酸化反応で分解されるDCA以外の基質に対して機能する.一方,2012年,Wandersらは,L-BPをコードする遺伝子EHHADH(enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase)のノックアウトマウスでは,極長鎖脂肪酸の分解は正常なものの,DCAの代謝産物であるアジピン酸(C6-DCA)やスベリン酸(C8-DCA)が産生されないことを見いだした38).すなわち,二頭酵素として同定されたL-BPとD-BPの基質特異性が異なり,L-BPは小胞体において脂肪酸のカルボキシ基とは反対に位置するω位のメチル基の水酸化によって生成されたω-ヒドロキシ脂肪酸がさらに酸化されたDCAの分解に機能することが明らかにされた.
4)3-ケトアシル-CoAチオラーゼとSCPx
β-酸化系の最終ステップは3-ケトアシル-CoAチオラーゼ(PTS2型輸送シグナル)あるいはSCPx(sterol carrier protein x, PTS1型)による3-ケトアシル-CoAのチオール開裂反応である.飽和および不飽和の極長鎖脂肪酸やDCA由来の3-ケトアシル-CoAはいずれの酵素によってもチオール開裂がなされる.一方,α-酸化反応によって分解されるプリスタン酸,DHCやTHC由来の3-ケトアシル-CoAのチオール開裂反応はSCPxが触媒する39).
ペルオキシソームのβ-酸化反応によって生じるH2O2はカタラーゼによって速やかに分解される.最近,著者らのグループはカタラーゼをはじめとするペルオキシソームマトリクスタンパク質の輸送障害性CHO変異細胞の一つであるZP114を用いてその相補遺伝子の探索を行ったところ,驚くべきことにミトコンドリア外膜タンパク質(ポリン)をコードするVDAC2(Voltage-Dependent Anion Channel 2)を同定した.詳細な解析の結果,VDAC2を受容体としてミトコンドリアに輸送され,アポトーシス促進因子として機能するBAKがVDAC2欠損によりペルオキシソームにも一部局在化し,カタラーゼのペルオキシソームからサイトゾルへの放出に関与することを発見した.さらに,広く知られるミトコンドリアでのBAKによる細胞死亢進とは逆に,ペルオキシソーム局在性BAKの活性化は,カタラーゼの放出を介して抗酸化ストレス反応として作用するというアポトーシス制御機構を明らかにした40, 41).また,小脳のプルキンエ細胞では,カタラーゼのペルオキシソーム局在性が低下するとも報告されており42),カタラーゼの積極的なサイトゾルへの局在化によるH2O2の効率的な分解を介した細胞機能制御機構の存在が推察される.
ペルオキシソームは脂肪酸の酸化反応のみならず,エーテル結合型リン脂質であるプラスマローゲンの生合成が開始される細胞小器官でもある.プラスマローゲンは,ジアシル型リン脂質とは異なり,グリセロール骨格のsn-1位に長鎖アルコールがビニルエーテル結合を介して結合したリン脂質である.生体内には極性基にエタノールアミンを有するエタノールアミンプラスマローゲン(PlsEtn)やコリンを有するコリンプラスマローゲン(PlsCho)が主として存在する.いずれも生体内に広く分布し,とくに前者は脳や中枢神経系に,後者は心臓や骨格筋に多く存在するが,肝臓での存在量は少ない43).また,脳・中枢神経系などPlsEtnを多く含む組織では,PlsEtnはホスファチジルエタノールアミン(PE)と同じ程度の存在量を示す43).
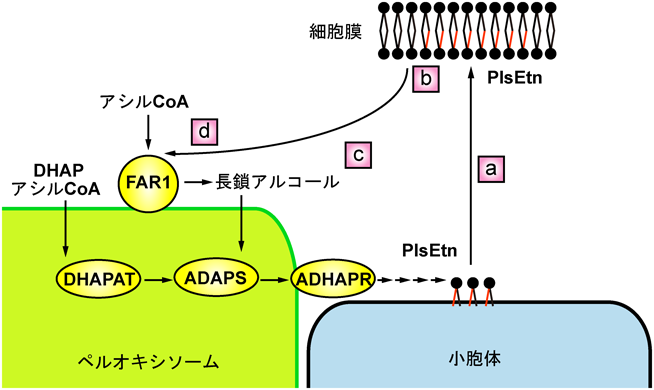
PlsEtnはペルオキシソームと小胞体に局在する酵素により全7段階の反応を経て合成される(図3).PlsEtnの合成は,ペルオキシソームマトリクスにおいてジヒドロキシアセトンリン酸(DHAP)のsn-1位にエーテル結合を介して長鎖アルコールが結合したアルキル-DHAPの形成がなされ,続いて小胞体においてsn-2, 3位それぞれに脂肪酸,エタノールアミンの付加により,アルキルアシル型リン脂質が合成された後,脱水素反応によりビニルエーテル結合が形成される44).PlsChoの生合成は,PlsEtnの塩基置換反応によって合成されると考えられているが,詳細な生合成経路は不明である.
プラスマローゲン生合成は,PTS1をC末端部に有するdihydroxyacetone phosphate acyltransferase(DHAPAT)によるアシル-DHAPのペルオキシソームマトリクスでの生合成で開始される.次いで,PTS2をN末端部に有するalkyl-dihydroxyacetone phosphate synthase(ADAPS)によってアシル-DHAPのアシル基は長鎖アルコールへと置換され,アルコールがエーテル結合を介してグリセロール骨格と結合したアルキル-DHAPが合成される.ADAPSの基質である長鎖アルコールは,ペルオキシソームのTA膜タンパク質であるfatty acyl-CoA reductase 1(FAR1)によって,C16およびC18のアシル-CoAが還元され産生される.FAR1には基質特異性が異なるFAR2が存在することが報告されている45).また,FAR1はさまざまな臓器で発現される一方,FAR2の発現は限定的である.最近明らかにされたFAR1欠損性患者の赤血球46)やDHAPATおよびADAPSの単独酵素機能障害性患者由来線維芽細胞47, 48)のプラスマローゲン量は,ペルオキシソーム欠損症患者の赤血球や患者由来線維芽細胞と同様に健常者細胞の約1/10程度まで著減することから,プラスマローゲン生合成においてこれら3種のペルオキシソーム局在性酵素が必須であることが示された.
プラスマローゲン合成不全は,関節点状石灰化,重度精神運動発達遅滞,白内障などを呈する肢根型点状軟骨異形成症(RCDP)を発症する43).これまでに,前述のDHAPAT, ADAPSおよびFAR1に加え,PEX7, PEX5Lを相補遺伝子とする5種の相補性群に分類されるRCDPが同定されている49, 50).PEX7, PEX5Lの遺伝子産物であるPex7とPex5Lは,いずれもPTS2を有するADAPSの細胞質からペルオキシソームマトリクスへの移行を担う輸送因子であることから6),これら因子の機能障害はADAPSのペルオキシソーム局在化障害によってプラスマローゲン合成不全を呈するものと推察される.
7. エタノールアミンプラスマローゲンの生合成制御
PlsEtnの生合成制御機構は不明であったが,著者らはプラスマローゲン合成不全変異細胞で数倍に上昇したFAR1の酵素活性および発現量は,野生型細胞と同レベルまでプラスマローゲン量を回復させるとFAR1の分解が亢進され野生型細胞と同程度までFAR1の発現量が低下することを見いだした51).さらに,野生型細胞のプラスマローゲン量の増加はFAR1の分解を促進しPlsEtnの生合成を抑制することも明らかにした52, 53).また,FAR1が合成する長鎖アルコールによるFAR1の活性阻害,すなわち生成物による活性の阻害はなされないとの知見54)から,PlsEtnの生合成は細胞内PlsEtn量を感知するフィードバック機構によるFAR1の分解制御で調節されると考えられる(図3)55).加えて,ペルオキシソーム欠損性マウスの腎臓においてFAR1の発現量が野生型マウスと比較して増加することも見いだされており56),個体においても細胞レベルと同様にPlsEtnの生合成は制御されるものと推察される.
最近,著者らはPlsEtnの生合成制御の重要なステップであるPlsEtnのセンシングに関し,細胞膜におけるPlsEtnが感知される知見を見いだした53).PlsEtnは小胞体でのビニルエーテル結合の形成によってその生合成が完了したのち,分泌経路非依存的に細胞膜を含むポスト-ゴルジ領域に輸送される57).細胞膜のコレステロールおよびスフィンゴミエリンに富むラフト画分の標識タンパク質であるグリコシルホスファチジルイノシトール(GPI)アンカータンパク質などのエンドサイトーシスに機能するFlotillin1の発現抑制は,ラフト画分に濃縮されるPlsEtnの量を増加させたことから,細胞膜におけるPlsEtnの一過的な増加の感知によりFAR1の分解が促進されたものと推察された53).さらに,PlsEtnの細胞膜内葉(inner leaflet)への偏在化を障害するフリッパーゼの機能阻害は,PlsEtnの細胞膜外葉(outer leaflet)への局在を増加させ,FAR1の分解を抑制することも見いだし,細胞膜におけるPlsEtnのセンシングが強く示唆された53).すなわち,PlsEtnの生合成は細胞膜内葉におけるPlsEtnの感知,その情報のペルオキシソームへの伝達,ペルオキシソーム膜におけるFAR1の分解調節で構成される高度な時空間的な機構で制御されるものと考えられる55).今後,PlsEtnのセンシングを担う分子の同定など,各ステップを担う因子の同定と機能解明が待たれる.
8. PlsEtnの恒常性依存的なコレステロールの生合成制御とその生理的意義
プラスマローゲンの生理機能は,多価不飽和脂肪酸の酸化に対する抗酸化作用,輸送小胞の融合促進などが考えられている43)が,具体的な機能はいまだ不明である.プラスマローゲンのsn-2位には,DHAやアラキドン酸(AA, C20:4n-6)などの多価不飽和脂肪酸が多く存在することから58)プラスマローゲンによる多価不飽和脂肪酸の貯蔵も示唆されている.興味深いことにプラスマローゲン欠損性患者由来の線維芽細胞やDHAPATノックアウトマウスの脳では,エタノールアミン含有性リン脂質(PEとPlsEtn)の総量は一定であるが,野生型マウスの脳と比較してAAを含むPEが増加したことから,プラスマローゲンは脳におけるDHAとAAの組成比の維持に寄与することが示唆された59).また,ADAPS機能障害性患者ではプラスマローゲン合成不全によるミエリン形成異常が報告されている60).DHAPATノックアウトマウスでは,ミエリン形成障害に加えて,視神経形成異常,精子形成異常,血液-精巣関門形態異常なども報告されている61, 62).
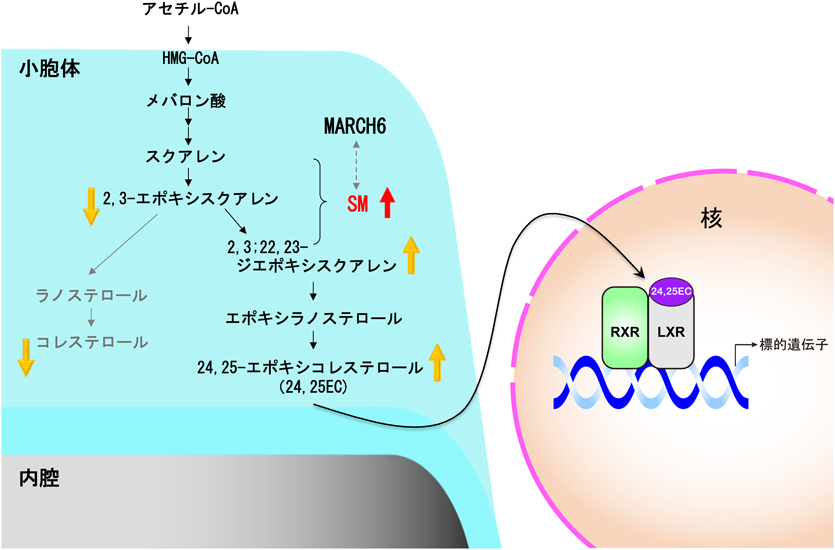
一方,プラスマローゲン欠損性変異細胞を用いた研究により,プラスマローゲン欠損によるHDL依存的なコレステロールの分泌阻害63),培地由来のコレステロールのエステル化64)やコレステロール生合成の抑制65)などコレステロールの恒常性との関連が見いだされた.プラスマローゲン欠損細胞におけるコレステロールの生合成の抑制は,小胞体に局在するコレステロール生合成の第2律速酵素であるスクアレンモノオキシゲナーゼ(squalene monooxygenase:SM)の安定化による24,25-エポキシコレステロール(24,25EC)の合成促進とコレステロールの生合成の抑制であることも明らかにしている65)(図4).24,25ECは核内受容体であるliver X receptor(LXR)の肝臓における主要なリガンドであることから,LXR依存的なコレステロール逆輸送系(reverse cholesterol transport)を効率的に維持させるために肝臓におけるPlsEtnの量は少なく維持されるものと推察される.
また,GPIを介して細胞膜に局在する哺乳類の赤血球アセチルコリンエステラーゼなどのGPIアンカー型タンパク質の脂肪酸の多くは1-アルキル-2-アシル型であるが,プラスマローゲン欠損性変異細胞ではジアシル型のGPIアンカーのみが検出されることが示され,GPIアンカータンパク質の生合成にもペルオキシソームのプラスマローゲン合成経路が重要とする新知見が示された66).
ペルオキシソームは,小胞体やミトコンドリアの代謝反応との機能的な連携による脂肪酸β-酸化反応等を介した異化・同化作用を展開することで,細胞機能を支えている.これらの代謝反応産物は,ミトコンドリアと小胞体の間で形成されるmitochondria-associated ER membrane(MAM)などのようなコンタクトサイトを介して効率的に運搬されることが想定される.すでに,酵母ペルオキシソームの分裂に関わるPex11を介したミトコンドリアとの接合67),哺乳類ペルオキシソームのACBD5を介した小胞体との接合などが明らかにされている68, 69).また,ペルオキシソームはミトコンドリアとともにRNAウイルスの侵入に対する免疫応答の惹起70)や脂肪滴とのつながりも示唆されている71).その他,Pex3, Pex19依存的な脂肪滴形成に関わるUBXD8の小胞体サブドメインへの標的化72),ミトコンドリアおよび小胞体由来小胞の融合を介するペルオキシソームのde novo合成経路73),Pex11β欠損による表皮前駆細胞の分裂面決定障害など74),ペルオキシソーム以外の細胞小器官との関連が種々見いだされつつある.今後,ペルオキシソームの恒常性維持に加え,他の細胞小器官との協調による機能制御を踏まえた研究の取り組みと進展は,ペルオキシソーム機能の理解をより深めるものと期待される.
引用文献References
1) de Duve, C. & Baudhuin, P. (1966) Physiol. Rev., 46, 323–357.
2) Lazarow, P.B. & de Duve, C. (1976) Proc. Natl. Acad. Sci. USA, 73, 2043–2046.
3) Brown, F.R. III, McAdams, A.J., Cummins, J.W., Konkol, R., Singh, I., Moser, A.B., & Moser, H.W. (1982) Johns Hopkins Med. J., 151, 344–351.
4) Singh, I., Moser, A.E., Goldfischer, S., & Moser, H.W. (1984) Proc. Natl. Acad. Sci. USA, 81, 4203–4207.
5) Heymans, H.S.A., Schutgens, R.B.H., Tan, R., van den Bosch, H., & Borst, P. (1983) Nature, 306, 69–70.
6) Fujiki, Y., Okumoto, K., Kinoshita, N., & Ghaedi, K. (2006) Biochim. Biophys. Acta, 1763, 1374–1381.
7) Ebberink, M.S., Koster, J., Visser, G., van Spronsen, F., Stolte-Dijkstra, I., Smit, G.P.A., Fock, J.M., Kemp, S., Wanders, R.J.A., & Waterham, H.R. (2012) J. Med. Genet., 49, 307–313.
8) Van Veldhoven, P.P. (2010) J. Lipid Res., 51, 2863–2895.
9) Wanders, R.J.A. & Waterham, H.R. (2006) Biochim. Biophys. Acta, 1763, 1707–1720.
10) Lazarow, P.B. & Fujiki, Y. (1985) Annu. Rev. Cell Biol., 1, 489–530.
11) Fujiki, Y., Okumoto, K., Mukai, S., Honsho, M., & Tamura, S. (2014) Front. Physiol., 5, 307.
12) Fujiki, Y. (2016) Proc. Jpn. Acad. Ser. B, 92, 463–477.
13) Okumoto, K., Misono, S., Miyata, N., Matsumoto, Y., Mukai, S., & Fujiki, Y. (2011) Traffic, 12, 1067–1083.
14) Miyata, N. & Fujiki, Y. (2005) Mol. Cell. Biol., 25, 10822–10832.
15) Matsumoto, N., Tamura, S., & Fujiki, Y. (2003) Nat. Cell Biol., 5, 454–460.
16) Matsuzono, Y., Matsuzaki, T., & Fujiki, Y. (2006) J. Cell Sci., 119, 3539–3550.
17) Yagita, Y., Hiromasa, T., & Fujiki, Y. (2013) J. Cell Biol., 200, 651–666.
18) Liu, Y., Yagita, Y., & Fujiki, Y. (2016) Traffic, 17, 433–455.
19) Matsuzaki, T. & Fujiki, Y. (2008) J. Cell Biol., 183, 1275–1286.
20) Schuldiner, M., Metz, J., Schmid, V., Denic, V., Rakwalska, M., Schmitt, H.D., Schwappach, B., & Weissman, J.S. (2008) Cell, 134, 634–645.
21) Lam, S.K., Yoda, N., & Schekman, R. (2010) Proc. Natl. Acad. Sci. USA, 107, 21523–21528.
22) Itoyama, A., Honsho, M., Abe, Y., Moser, A.B., Yoshida, Y., & Fujiki, Y. (2012) J. Cell Sci., 125, 589–602.
23) Tanaka, A., Kobayashi, S., & Fujiki, Y. (2006) Exp. Cell Res., 312, 1671–1684.
24) Kim, P.K., Hailey, D.W., Mullen, R.T., & Lippincott-Schwartz, J. (2008) Proc. Natl. Acad. Sci. USA, 105, 20567–20574.
25) Narendra, D., Tanaka, A., Suen, D.-F., & Youle, R.J. (2008) J. Cell Biol., 183, 795–803.
26) Yamashita, S., Abe, K., Tatemichi, Y., & Fujiki, Y. (2014) Autophagy, 10, 1549–1564.
27) Zhang, J., Tripathi, D.N., Jing, J., Alexander, A., Kim, J., Powell, R.T., Dere, R., Tait-Mulder, J., Lee, J.-H., Paull, T.T., Pandita, R.K., Charaka, V.K., Pandita, T.K., Kastan, M.B., & Walker, C.L. (2015) Nat. Cell Biol., 17, 1259–1269.
28) Sargent, G., van Zutphen, T., Shatseva, T., Zhang, L., Di Giovanni, V., Bandsma, R., & Kim, P.K. (2016) J. Cell Biol., 214, 677–690.
29) Baker, A., Carrier, D.J., Schaedler, T., Waterham, H.R., van Roermund, C.W., & Theodoulou, F.L. (2015) Biochem. Soc. Trans., 43, 959–965.
30) Geillon, F., Gondcaille, C., Charbonnier, S., Van Roermund, C.W., Lopez, T.E., Dias, A.M.M., Pais de Barros, J.-P., Arnould, C., Wanders, R.J.A., Trompier, D., & Savary, S. (2014) J. Biol. Chem., 289, 24511–24520.
31) Yagita, Y., Shinohara, K., Abe, Y., Nakagawa, K., Al-Owain, M., Alkuraya, F.S., & Fujiki, Y. (2017) J. Biol. Chem., 292, 691–705.
32) Ferdinandusse, S., Falkenberg, K.D., Koster, J., Mooyer, P.A., Jones, R., van Roermund, C.W.T., Pizzino, A., Schrader, M., Wanders, R.J.A., Vanderver, A., & Waterham, H.R. (2017) J. Med. Genet., 54, 330–337.
33) Costello, J.L., Castro, I.G., Schrader, T.A., Islinger, M., & Schrader, M. (2017) Cell Cycle, 16, 1039–1045.
34) Mosser, J., Douar, A.-M., Sarde, C.-O., Kioschis, P., Feil, R., Moser, H., Poustka, A.-M., Mandel, J.-L., & Aubourg, P. (1993) Nature, 361, 726–730.
35) Ferdinandusse, S., Jimenez-Sanchez, G., Koster, J., Denis, S., Van Roermund, C.W., Silva-Zolezzi, I., Moser, A.B., Visser, W.F., Gulluoglu, M., Durmaz, O., Demirkol, M., Waterham, H.R., Gökcay, G., Wanders, R.J.A., & Valle, D. (2015) Hum. Mol. Genet., 24, 361–370.
36) Kurochkin, I.V., Mizuno, Y., Konagaya, A., Sakaki, Y., Schönbach, C., & Okazaki, Y. (2007) EMBO J., 26, 835–845.
37) Okumoto, K., Kametani, Y., & Fujiki, Y. (2011) J. Biol. Chem., 286, 44367–44379.
38) Houten, S.M., Denis, S., Argmann, C.A., Jia, Y., Ferdinandusse, S., Reddy, J.K., & Wanders, R.J.A. (2012) J. Lipid Res., 53, 1296–1303.
39) Wanders, R.J.A., Denis, S., van Berkel, E., Wouters, F., Wirtz, K.W.A., & Seedorf, U. (1998) J. Inherit. Metab. Dis., 21, 302–305.
40) Hosoi, K., Miyata, N., Mukai, S., Furuki, S., Okumoto, K., Cheng, E.H., & Fujiki, Y. (2017) J. Cell Biol., 216, 709–721.
41) Fujiki, Y., Miyata, N., Mukai, S., Okumoto, K., & Cheng, E.H. (2017) Mol. Cell. Oncol., 4, e1306610.
42) Nagase, T., Shimozawa, N., Takemoto, Y., Suzuki, Y., Komori, M., & Kondo, N. (2004) Biochim. Biophys. Acta, 1671, 26–33.
43) Braverman, N.E. & Moser, A.B. (2012) Biochim. Biophys. Acta, 1822, 1442–1452.
44) Nagan, N. & Zoeller, R.A. (2001) Prog. Lipid Res., 40, 199–229.
45) Cheng, J.B. & Russell, D.W. (2004) J. Biol. Chem., 279, 37789–37797.
46) Buchert, R., Tawamie, H., Smith, C., Uebe, S., Innes, A.M., Al Hallak, B., Ekici, A.B., Sticht, H., Schwarze, B., Lamont, R.E., Parboosingh, J.S., Bernier, F.P., & Abou Jamra, R. (2014) Am. J. Hum. Genet., 95, 602–610.
47) Wanders, R.J.A., Schumacher, H., Heikoop, J., Schutgens, R.B.H., & Tager, J.M. (1992) J. Inherit. Metab. Dis., 15, 389–391.
48) Wanders, R.J.A., Dekker, C., Hovarth, V.A., Schutgens, R.B., Tager, J.M., van Laer, P., & Lecoutere, D. (1994) J. Inherit. Metab. Dis., 17, 315–318.
49) Braverman, N., Steel, G., Obie, C., Moser, A., Moser, H., Gould, S.J., & Valle, D. (1997) Nat. Genet., 15, 369–376.
50) Barøy, T., Koster, J., Strømme, P., Ebberink, M.S., Misceo, D., Ferdinandusse, S., Holmgren, A., Hughes, T., Merckoll, E., Westvik, J., Woldseth, B., Walter, J., Wood, N., Tvedt, B., Stadskleiv, K., Wanders, R.J.A., Waterham, H.R., & Frengen, E. (2015) Hum. Mol. Genet., 24, 5845–5854.
51) Honsho, M., Asaoku, S., & Fujiki, Y. (2010) J. Biol. Chem., 285, 8537–8542.
52) Honsho, M., Asaoku, S., Fukumoto, K., & Fujiki, Y. (2013) J. Biol. Chem., 288, 34588–34598.
53) Honsho, M., Abe, Y., & Fujiki, Y. (2017) Sci. Rep., 7, 43936.
54) Bishop, J.E. & Hajra, A.K. (1981) J. Biol. Chem., 256, 9542–9550.
55) Honsho, M. & Fujiki, Y. (2017) FEBS Lett., 591, 2720–2729.
56) Wiese, S., Gronemeyer, T., Brites, P., Ofman, R., Bunse, C., Renz, C., Meyer, H.E., Wanders, R.J.A., & Warscheid, B. (2012) Int. J. Mass Spectrom., 312, 30–40.
57) Honsho, M., Yagita, Y., Kinoshita, N., & Fujiki, Y. (2008) Biochim. Biophys. Acta, 1783, 1857–1865.
58) Abe, Y., Honsho, M., Nakanishi, H., Taguchi, R., & Fujiki, Y. (2014) Biochim. Biophys. Acta, 1841, 610–619.
59) Dorninger, F., Brodde, A., Braverman, N.E., Moser, A.B., Just, W.W., Forss-Petter, S., Brügger, B., & Berger, J. (2015) Biochim. Biophys. Acta, 1851, 117–128.
60) Sztriha, L., Al-Gazali, L.I., Wanders, R.J.A., Ofman, R., Nork, M., & Lestringant, G.G. (2000) Dev. Med. Child Neurol., 42, 492–495.
61) Teigler, A., Komljenovic, D., Draguhn, A., Gorgas, K., & Just, W.W. (2009) Hum. Mol. Genet., 18, 1897–1908.
62) Komljenovic, D., Sandhoff, R., Teigler, A., Heid, H., Just, W.W., & Gorgas, K. (2009) Cell Tissue Res., 337, 281–299.
63) Mandel, H., Sharf, R., Berant, M., Wanders, R.J.A., Vreken, P., & Aviram, M. (1998) Biochem. Biophys. Res. Commun., 250, 369–373.
64) Munn, N.J., Arnio, E., Liu, D., Zoeller, R.A., & Liscum, L. (2003) J. Lipid Res., 44, 182–192.
65) Honsho, M., Abe, Y., & Fujiki, Y. (2015) J. Biol. Chem., 290, 28822–28833.
66) Kanzawa, N., Maeda, Y., Ogiso, H., Murakami, Y., Taguchi, R., & Kinoshita, T. (2009) Proc. Natl. Acad. Sci. USA, 106, 17711–17716.
67) Mattiazzi Ušaj, M., Brložnik, M., Kaferle, P., Žitnik, M., Wolinski, H., Leitner, F., Kohlwein, S.D., Zupan, B., & Petrovič, U. (2015) J. Mol. Biol., 427, 2072–2087.
68) Costello, J.L., Castro, I.G., Hacker, C., Schrader, T.A., Metz, J., Zeuschner, D., Azadi, A.S., Godinho, L.F., Costina, V., Findeisen, P., Manner, A., Islinger, M., & Schrader, M. (2017) J. Cell Biol., 216, 331–342.
69) Hua, R., Cheng, D., Coyaud, É., Freeman, S., Di Pietro, E., Wang, Y., Vissa, A., Yip, C.M., Fairn, G.D., Braverman, N., Brumell, J.H., Trimble, W.S., Raught, B., & Kim, P.K. (2017) J. Cell Biol., 216, 367–377.
70) Dixit, E., Boulant, S., Zhang, Y., Lee, A.S., Odendall, C., Shum, B., Hacohen, N., Chen, Z.J., Whelan, S.P., Fransen, M., Nibert, M.L., Superti-Furga, G., & Kagan, J.C. (2010) Cell, 141, 668–681.
71) Schrader, M. (2001) J. Histochem. Cytochem., 49, 1421–1429.
72) Schrul, B. & Kopito, R.R. (2016) Nat. Cell Biol., 18, 740–751.
73) Sugiura, A., Mattie, S., Prudent, J., & McBride, H.M. (2017) Nature, 542, 251–254.
74) Asare, A., Levorse, J., & Fuchs, E. (2017) Science, 355, eaah4701.
75) Waterham, H.R., Ferdinandusse, S., & Wanders, R.J.A. (2016) Biochim. Biophys. Acta, 1863, 922–933.
著者紹介Author Profile
 本庄 雅則(ほんしょう まさのり)
本庄 雅則(ほんしょう まさのり)九州大学生体防御医学研究所特任准教授.理学博士.
略歴1992年九州大学理学部卒業.97年同大学院理学研究科化学専攻博士後期課程修了.マックス・プランク分子細胞生物学・遺伝学研究所,九州大学大学院理学研究院生物科学部門特任助教,准教授などを経て2015年より現職.
研究テーマと抱負ペルオキシソームとその代謝産物(とくにエーテルリン脂質プラスマローゲン)の生理機能に関し,細胞や個体において緻密に制御される恒常性維持機構を含めて明らかにしたい.
ウェブサイトhttp://www.organelle.kyushu-u.ac.jp/lab/organellestasis/index.html
趣味テニス,1歳半の息子の高齢育児.
 藤木 幸夫(ふじき ゆきお)
藤木 幸夫(ふじき ゆきお)九州大学生体防御医学研究所特任教授.農学博士.
略歴1976年九州大学大学院農学研究科博士課程修了(農学博士).76年コーネル大学博士研究員.79年ロックフェラー大学上級研究員,助教授.85年明治乳業(株)ヘルスサイエンス研究所主任研究員,研究室長.94年九州大学理学部教授.2015年より現職.
研究テーマと抱負タンパク質の細胞内選別輸送:細胞小器官ペルオキシソームの形成制御の分子機構およびその障害・病態の発現機構の研究により,オルガネラの恒常性と細胞機能制御の課題を明らかにしたい.
ウェブサイトhttp://www.organelle.kyushu-u.ac.jp/lab/organellestasis/index.html