我々の細胞にはミトコンドリアとペルオキシソームという二つの脂肪酸酸化に関わる小器官(オルガネラ)がある.脂肪酸をβ酸化するという働きにおいて両者は共通であるが,役割は異なる.たとえば,ミトコンドリアの場合,脂肪酸β酸化によって得られるアセチルCoA, FADH2およびNADHはミトコンドリアマトリックスのクエン酸回路と内膜上の電子伝達系に供され,ATP産生に利用される.ミトコンドリアにおける脂肪酸酸化の一義的な役割はエネルギー産生である.一方,ペルオキシソームにはそのようなATP産生機能がなく,脂肪酸の酸化反応によって生じるアセチルCoAやNADHは直接ATP産生に利用されない.すなわち,ミトコンドリアほどエネルギー産生との関係を持たない.ぺルオキシソームの脂肪酸β酸化は多種多様な脂溶性物質を酸化分解するので,不要な脂溶性物質の水溶性物質への変換といった薬物代謝的意義があるが,脂質合成の側面もある.ドコサヘキサエン酸(DHA),プラスマローゲンおよび胆汁酸の生合成にはペルオキシソームが関与している.また,後述するようにペルオキシソームは脂肪酸の作り替えに役割を果たしている可能性がある.以下にペルオキシソームでβ酸化を受ける代表的な基質について述べる(図1).
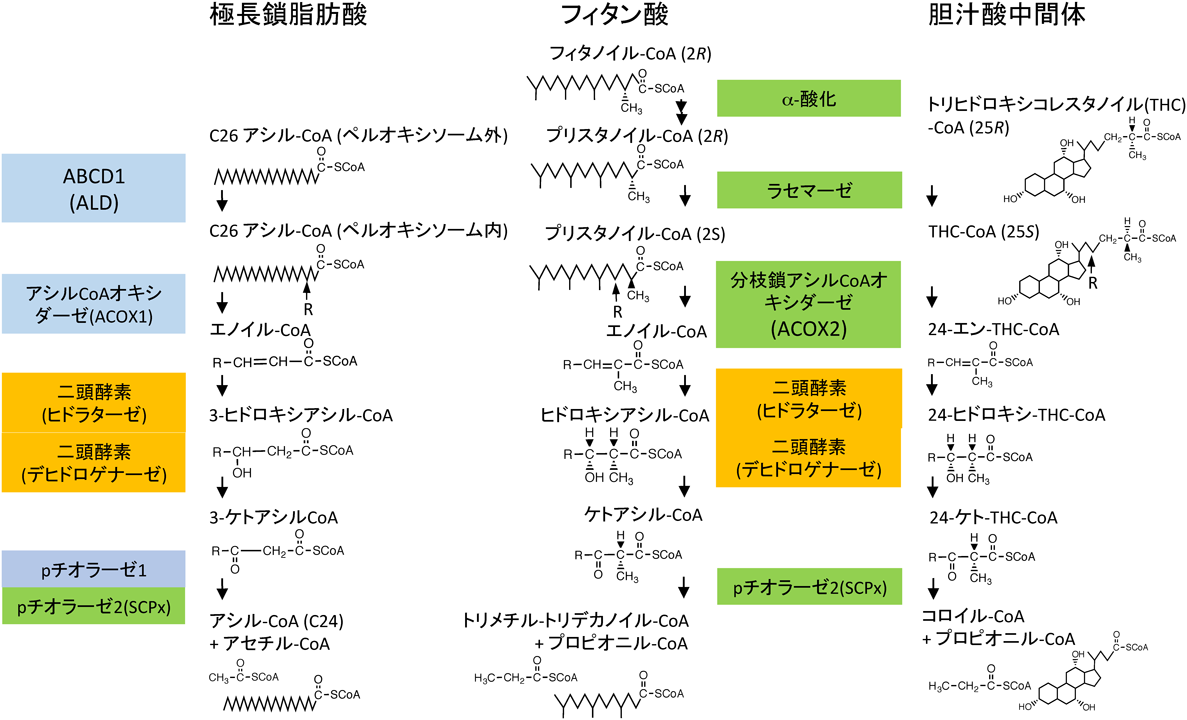
1)極長鎖脂肪酸
C24以上の脂肪酸はもっぱらペルオキシソームで酸化消去される.このことはZellweger症候群のようなペルオキシソーム形成異常症やALD, ACOX1欠損症,DBP欠損症のような脂肪酸β酸化反応に関わるタンパク質の欠損症でC24やC26といった極長鎖脂肪酸が全身性に蓄積することから明らかである.
2)2-メチル分枝脂肪酸/プリスタン酸
フィトールおよびフィタン酸はそれぞれ植物性油脂や反芻動物の脂肪および乳製品に含まれる分枝脂肪アルコールおよび分枝脂肪酸である.C20のフィタン酸はCoA体となった後,ペルオキシソームでフィタノイルCoAヒドロキシラーゼ(PhyH)が関わる反応でα酸化を受け,C19のプリスタン酸となる.その後,3サイクルのβ酸化を受ける2).PhyHはペルオキシソーム酵素でありこれをコードする遺伝子はレフサム病の原因遺伝子である.
3)胆汁酸前駆体(ジ/トリヒドロキシコレスタン酸)
代表的な一次胆汁酸であるコール酸やケノデオキシコール酸はコレステロールから合成されるが,直鎖状炭化水素鎖の鎖長短縮にはまず,ジ/トリヒドロキシコレスタン酸(D/THCA)の25位のメチル基のRをSに変換するα-メチルアシルCoAラセマーゼ(AMACR)が働き3),次いで,ペルオキシソームのβ酸化1サイクルで鎖長短縮される.
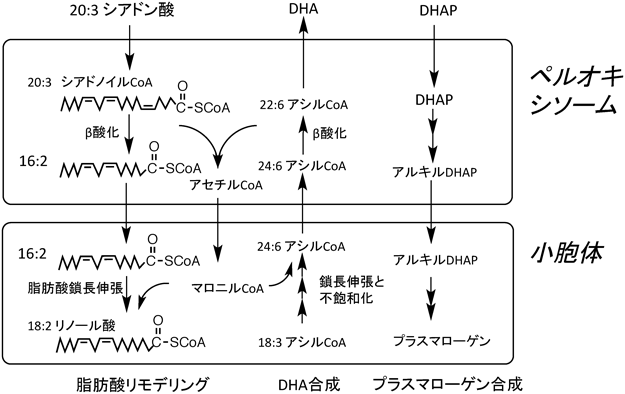
4)ドコサヘキサエン酸(DHA)
DHAの生合成にはΔ4不飽和化酵素活性が関わると考えられてきたが,長らくその実態は同定されなかった.現在,DHA(Δ-4,7,10,13,16,19)は24 : 6(Δ-6,9,12,15,18,21)がペルオキシソームのβ酸化1サイクルによって鎖長短縮されることで生合成されることが明らかとされている4)(図2).
5) ポリメチレン中断型不飽和脂肪酸
植物にはポリメチレンで中断された二重結合を有する多価不飽和脂肪酸が存在する.ポリメチレン中断型不飽和脂肪酸はペルオキシソームでの鎖長短縮とミクロソームでの鎖長伸張を受け,リノール酸やリノレン酸に変換される5).
3. ペルオキシソームβ酸化代謝物の脂肪酸合成への利用
哺乳類の持つ多価不飽和脂肪酸の二重結合配置はメチレン中断型である.これに対し,裸子植物にはポリメチレンで中断された二重結合配置を持つ多価不飽和脂肪酸が含まれている6).代表的ポリメチレン中断型脂肪酸はシアドン酸20 : 3(Δ-5,11,14)とジュニペロン酸20 : 4(Δ-5,11,14,17)で,食事性に摂取している脂肪酸でもある.我々はシアドン酸が動物細胞内においてペルオキシソームの2サイクルβ酸化で16 : 2(Δ-7,10)に鎖長短縮され,ミクロソームの鎖長伸張系にてリノール酸へと変換されることを見いだした(図2).この代謝はペルオキシソーム欠損CHO細胞(PEX5欠損)では起きない.また,n-3系列のC20ポリメチレン中断型不飽和脂肪酸であるジュニペロン酸,20 : 4(Δ-8,11,14,17)はシアドン酸と同様に代謝され,α-リノレン酸に変換されることも確認している5).ポリメチレン中断型不飽和脂肪酸は高等動物の脂質ではみられない多価不飽和脂肪酸である.我々は食事性に摂取したポリメチレン中断型不飽和脂肪酸を排除するが,そのやり方はペルオキシソームとミトコンドリアで完全に酸化分解するのではない.ペルオキシソームβ酸化系で部分的に鎖長短縮し,ミクロソームで鎖長伸張を行うことで,我々の細胞に適した必須脂肪酸に作り替えている.この脂肪酸リモデリング代謝系はヒトにも備わっている.たとえば,ヒト胃がん由来MKN74細胞では細胞内に取り込まれたシアドン酸の50%が24時間でリノール酸に変換されており7),主要代謝経路として機能している.さらに,ペルオキシソームで生じるアセチルCoAはマロニルCoAに変換され,長鎖脂肪酸の鎖長伸張に利用されると報告されている8, 9).すなわち,ペルオキシソームの脂肪酸酸化は長鎖脂肪酸生合成のための材料供給を行っている可能性がある.ペルオキシソームは脂肪酸生合成系に近いところに立ち位置があるのかもしれない.
最近,ペルオキシソーム内で生じたβ酸化産物をペルオキシソーム外に排出する様式がわかってきた.アセチルCoAや鎖長短縮されたアシルCoAは対応するアシル(アセチル)カルニチンに変換されるか,チオエステラーゼで遊離脂肪酸(酢酸)に変換されることでペルオキシソーム膜を横切る可能性が提唱されている10).
4. ペルオキシソームにおける脂肪酸酸化酵素の遺伝的欠損と疾患
1)ペルオキシソーム病の分類
ペルオキシソームの成熟にはペルオキシソームに局在する酵素やタンパク質を特異的に中に運び入れることが必要である.このプロセスにはペルオキシソームタンパク質の輸送シグナル配列を認識する受容体タンパク質,ペルオキシソームタンパク質とその受容体からなる複合体と結合するドッキングタンパク質,そのリサイクリングに関わるタンパク質など多くのタンパク質が関わっている.このようなタンパク質とこれをコードする遺伝子はPEXタンパク質/遺伝子と呼ばれ,ヒトでは14種が知られている11).このうちペルオキシソームの形成やタンパク質の輸送に関わる13種類のPEX遺伝子がペルオキシソーム形成異常症として分類されている.著者らは最初に病因として報告されたPEX212)をはじめとして,いくつかのPEX遺伝子がヒトの病因であることを明らかにし,2004年には13番目の病因遺伝子としてPEX14遺伝子異常を報告している13).その後,2012年にはペルオキシソームの分裂に関わるPEX11βの遺伝子異常患者が報告されている14).
ペルオキシソーム形成異常症の代表的疾患はZellweger症候群で,出生時の筋緊張低下,哺乳障害,痙攣,異常顔貌(前額突出,鼻根部扁平),脳室拡大,肝腫大を呈し,国内診断例の生存期間は2~14か月である11).この疾患の生化学的特徴としては先にあげたペルオキシソームβ酸化の基質である極長鎖脂肪酸やプリスタン酸とその上流のフィタン酸,胆汁酸中間代謝物の蓄積を認め,さらにその生合成にペルオキシソームが関わるプラスマローゲンの低下も認める11).ペルオキシソーム形成異常症にはZellweger症候群以外に重症度の低い新生児型副腎白質ジストロフィーや乳児型レフサム病,骨系統疾患に属し受容体タンパク質のPEX7やPEX5 long isoformの遺伝子異常が判明した根性点状軟骨異形成症1型や5型15)が分類されている.生化学的には前者の患者ではZellweger症候群に比べて軽度の異常がみられ,後者のPEX7やPEX5 long isoformの遺伝子異常患者では認識するタンパク質の機能を反映してフィタン酸の蓄積とプラスマローゲンの低下を認めるが,極長鎖脂肪酸の蓄積はみられない.一方,ペルオキシソームの分裂に関わるPEX11β遺伝子異常では,報告されているナンセンス変異をホモ接合で有する患者においては形態的にペルオキシソームの分裂異常を認めるものの,形成異常症患者でみられる代謝異常はみられない14).
ペルオキシソームに局在する酵素やトランスポーター自体の遺伝子異常は単独酵素欠損症に分類される.ペルオキシソームに極長鎖脂肪酸CoAを特異的に通過させる膜タンパク質ABCD1(ALDタンパク質)や,脂肪酸β酸化反応を遂行するアシルCoAオキシダーゼ(ACOX1)や二頭酵素(DBP)など10種類以上の遺伝子異常症が知られ,中枢神経をはじめ多彩な臨床像を呈している.これらのペルオキシソーム病の病因,病態,治療と予後は参考文献11)に詳しい.次項ではペルオキシソームにおける脂肪酸β酸化が滞るとなぜ予後不良の神経変性が起こるのか,ペルオキシソーム形成異常症の病因であるPEX5遺伝子や脂肪酸β酸化に関わる酵素遺伝子のコンディショナルノックアウトマウスと生化学的異常から病態について文献的考察を加えた.
2)PEX5欠損
PEX5の重度の遺伝子異常によりその機能が失われると,ほとんどのペルオキシソームタンパク質はペルオキシソームに輸送されない.PEX5の機能欠損マウスはペルオキシソーム機能を欠落した空のペルオキシソーム膜構造物(ghost)しか持たない個体となり,ヒトでは最重症型のZellweger症候群を呈する11).
a. 全身PEX5欠損
PEX5欠損マウスは胎仔の段階での成長が遅く神経細胞の移動異常のために,正常な脳組織が形成されない.また,重篤な筋緊張低下を起こしており,出生後72時間以内に死亡する16).筋緊張低下や神経細胞の移動異常はヒトのZellweger症候群においてもみられる症状である.
b. 外胚葉由来神経系細胞のPEX5欠損
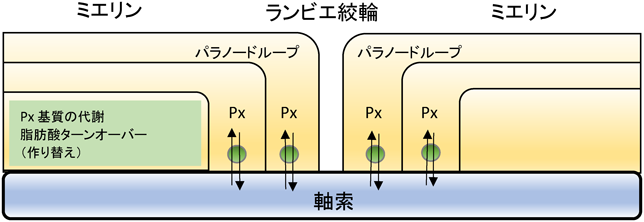
神経組織を形成する外胚葉由来の細胞には神経細胞,アストログリア,オリゴデンドロサイトがある.Nestin-Pex5欠損マウスは神経幹細胞特異的にPEX5を欠損するのでこれら3種の外胚葉由来神経系細胞すべてでペルオキシソームが欠損した個体となるが,ミクログリアや内皮細胞,線維芽細胞には影響を与えない.このNestin-Pex5欠損マウスは正常に出生するが,1週齢から成長の遅れが目立ち,プルキンエ細胞の層構造異常や,樹状突起が少なく分子層が薄いなどの小脳の形成異常を起こす.運動能力と認識能力が低下し,生存期間は6か月未満である17, 18).また,ミエリン形成異常もミエリン変性も2~3週齢から脳のすべての部位で劇的に起こる.脱髄の進行とともにミクログリアの活性化が起こり神経炎症を呈する19).
c. 神経細胞特異的(NEX-Pex5−/−),アストロサイト特異的(Gfap-Pex5−/−)およびオリゴデンドロサイト特異的(Cnp-Pex5−/−)PEX5欠損
神経細胞特異的PEX5欠損マウスは特に異常を示さず,脱髄も起こらない.グリア細胞の活性化も起こらず,マウスの平均寿命の2年間生存する20).アストロサイト特異的PEX5欠損マウスも極長鎖脂肪酸の蓄積はみられるものの明らかな神経症状は現れない21).これらに対し,オリゴデンドロサイト特異的PEX5欠損マウスは外胚葉由来神経細胞特異的PEX5欠損マウスと同様の神経症状を呈する22, 23).すなわち,マウスではオリゴデンドロサイトのペルオキシソームが神経組織の維持に最も重要な役割を果たしているということになる.神経症状を呈するPEX5欠損マウスは軸索腫張が先行して起こる21, 22).
3)ABCD1(ALD)欠損
ABCD1は以前にはALDタンパク質とも呼ばれ,ペルオキシソーム病の中で最も患者数の多い副腎白質ジストロフィー(adrenoleukodystrophy:ALD)の原因タンパク質である.ABCD1は極長鎖脂肪酸CoAのペルオキシソーム内への輸送をつかさどるATP依存性トランスポーターであり,このタンパク質を欠損すると極長鎖脂肪酸をペルオキシソームに輸送することができなくなるために,全身性にC24やC26といった極長鎖脂肪酸が蓄積する.このことは1976年,五十嵐らによりALD患者において発見され23),以後,血液中の極長鎖脂肪酸の蓄積がこの疾患の診断に利用されるようになった.ALD患者ではペルオキシソームのβ酸化基質であるフィタン酸や胆汁酸中間体の蓄積は認められないことより,この疾患のABCD1遺伝子異常による生化学的異常はABCD1がエントリーに関わる極長鎖脂肪酸が酸化できないことにあると考えられる.ALDには中枢神経障害を呈する小児大脳型(30~35%),思春期大脳型(4~9%),成人期大脳型(20%),小脳脳幹型(8%)の他,脊髄障害を呈する副腎脊髄ニューロパチー(adrenomyeloneuropathy:AMN)(25%)や副腎不全のみを来すAddison型など多彩な臨床型がある11).ABCD1遺伝子変異はすでに732種類がデータベースに掲載されている(http://www.x-ald.nl).この遺伝子変異と臨床型との間には相関がなく,多彩な臨床型はABCD1の遺伝子変異型では説明できない11).また,極長鎖脂肪酸の蓄積の程度と臨床型の重症度も相関しないため,大脳型病変部で観察される脱髄に極長鎖脂肪酸がどのように関わっているかも明確ではない11).一方,ABCD1の欠損マウスでは脳,脊髄,肺,腎での極長鎖脂肪酸の蓄積や線維芽細胞や肝細胞での極長鎖脂肪酸β酸化活性の低下は認めるものの,脳,脊髄,末梢神経での異常は認められなかった24, 25).しかし,その後15月齢以降に神経行動異常や神経伝達速度の遅延,脊髄や末梢神経の異常を起こすことが明らかとなった26).高齢期に出現するこの症状はヒトALDの臨床型のうちAMN症状と類似していることから,ヒトでもマウスでもABCD1欠損の単独要因によって引き起こされる基本病態はAMNと考えられている26).
このABCD1欠損マウスはAMNの症状を呈する前に活性酸素ストレスにさらされていると報告されている27).また,C26極長鎖脂肪酸とともにインキュベートしたオリゴデンドロサイトはミトコンドリアが正常に機能せず活性酸素による細胞死が起こることも示されている28).これらを背景にAMN患者への補助療法として抗酸化剤の有効性を調べる臨床試験が行われている29).
4)アシルCoAオキシダーゼ(ACOX)と二頭酵素(DBP)欠損
ACOX1は脂肪酸β酸化の最初の反応を触媒する酵素である(図1).ヒトで本酵素を欠損すると新生児期からの筋緊張低下,乳児期からの痙攣,視力・聴力障害となり2歳過ぎから退行して死亡する.生化学的特徴は飽和極長鎖脂肪酸の蓄積である.分枝鎖脂肪酸には別のACOX(ACOX2)が対応しているため,ACOX1欠損症ではフィタン酸,プリスタン酸,胆汁酸中間代謝物の蓄積はみられない11).ACOX1欠損マウスは成長の遅れ,ライディッヒ細胞の枯渇による不妊,極長鎖脂肪酸の蓄積を特徴とし,肝臓障害が顕著に観察されるものの重篤な神経変性には至らないようである30).
DBP欠損症はより重篤であり,ヒトもマウスも神経変性に至る.この酵素はACOXで生成したエノイル-CoAをD-3-ヒドロキシアシル-CoAに変換するヒドラターゼ活性,次いで,3-ケトアシル-CoAに酸化するデヒドロゲナーゼ活性の二つの反応を触媒する酵素で,二頭酵素(D-bifunctional protein:DBP)と呼ばれている(図1).この酵素は直鎖状の脂肪酸の酸化にも分枝脂肪酸の酸化にも関わるので,この酵素を欠損すると飽和極長鎖脂肪酸だけでなく,フィタン酸,プリスタン酸,胆汁酸中間代謝物が蓄積する11).この酵素を欠損した患者の場合,ほとんどが新生児期からの筋緊張低下がみられ,Zellweger症候群類似の病態となり,髄鞘化遅延,脱髄,小脳や脳梁の低形成がみられ,多くは2歳までに死亡する.ALDでは臨床型の重症度と極長鎖脂肪酸の蓄積量が相関しないが,二頭酵素欠損症の場合はペルオキシソームにおけるβ酸化基質の蓄積量と生存期間との明らかな相関がある.マウスでも二頭酵素を欠損すると極長鎖脂肪酸や分枝脂肪酸の蓄積が起こり重篤な症状を呈する.生まれた直後は異常を認めないものの,成長がひどく抑制され,1/3は離乳前に死亡し31),残りも運動障害を起こし半年以内に死亡する.また,灰白質と脊髄にアストログリアや反応性ミクログリアの増殖がみられ,小脳と脳幹に脱髄がみられる.この欠損動物にフィトールを摂餌しても悪化しないことから,分枝脂肪酸の蓄積は直接の原因ではないと考えられる32).
引用文献References
1) Goldfischer, S., Moore, C.L., Johnson, A.B., Spiro, A.J., Valsamis, M.P., Wisniewski, H.K., Ritch, R.H., Norton, W.T., Rapin, I., & Gartner, L.M. (1973) Science, 182, 62–64.
2) Baes, M. & Van Veldhoven, P.P. (2012) Biochim. Biophys. Acta, 1822, 1489–1500.
3) Ferdinandusse, S. & Houten, S.M. (2006) Biochim. Biophys. Acta, 1763, 1427–1440.
4) Voss, A., Reinhart, M., Sankarappa, S., & Sprecher, H. (1991) J. Biol. Chem., 266, 19995–20000.
5) Tanaka, T., Morishige, J., Iwawaki, D., Fukuhara, T., Hamamura, N., Hirano, K., Osumi, T., & Satouchi, K. (2007) FEBS J., 274, 2728–2737.
6) Wolff, R.L., Deluc, L.G., & Marpeau, A.M. (1996) J. Am. Oil Chem. Soc., 73, 765–771.
7) Tanaka, T., Uozumi, S., Morito, K., Osumi, T., & Tokumura, A. (2014) Lipids, 49, 423–429.
8) Wong, D.A., Bassilian, S., Lim, S., & Lee, W.-N.P. (2004) J. Biol. Chem., 279, 41302–41309.
9) Reszko, A.E., Kasumov, T., David, F., Jobbins, K.A., Thomas, K.R., Hoppel, C.L., Brunengraber, H., & Rosiers, C.D. (2004) J. Biol. Chem., 279, 19574–19579.
10) Antonenkov, V.D. & Hiltunen, J.K. (2012) Biochim. Biophys. Acta, 1822, 1374–1386.
11) 下澤伸行(2013)ペルオキシソーム病ハンドブック2013,日本臨牀社.
12) Shimozawa, N., Tsukamoto, T., Suzuki, Y., Orii, T., Shirayosi, Y., Mori, T., & Fujilo, Y. (1992) Science, 255, 1132–1134.
13) Shimozawa, N., Tsukamoto, T., Nagase, T., Takemoto, Y., Koyama, N., Suzuki, Y., Komori, M., Osumi, T., Jeannette, G., Wanders, R.J.A., & Kondo, N. (2004) Hum. Mutat., 23, 552–558.
14) Ebberink, M.S., Koster, J., Visser, G., Spronsen, F., Stolte-Dijkstra, I., Smit, G.P., Fock, J.M., Kemp, S., Wanders, R.J.A., & Waterham, H.R. (2012) J. Med. Genet., 49, 307–313.
15) Barøy, T., Koster, J., Strømme, P., Ebberink, M.S., Misceo, D., Ferdinandusse, S., Holmgren, A., Hughes, T., Merckoll, E., Westvik, J., Woldseth, B., Walter, J., Wood, N., Tvedt, B., Stadskleiv, K., Wanders, R.J., Waterham, H.R., & Frengen, E. (2015) Hum. Mol. Genet., 24, 5845–5854.
16) Baes, M., Gressens, P., Baumgart, E., Carmeliet, P., Casteels, M., Fransen, M., Evrard, P., Fahimi, D., Declercq, P.E., Collen, D., Van Veldhoven, P., & Mannaerts, G.P. (1997) Nat. Genet., 17, 49–57.
17) Krysko, O., Hulshagen, L., Janssen, A., Schüts, G., Klein, R., De Bruycker, M., Espeel, M., Gressens, P., & Baes, M. (2007) J. Neurosci. Res., 85, 58–72.
18) Hulshagen, L., Krysko, O., Bottelbergs, A., Huyghe, S., Klein, R., Van Veldhoven, P.P., De Deyn, P.P., D’Hooge, R., Hartmann, D., & Baes, M. (2008) J. Neurosci., 28, 4015–4027.
19) Bottelbergs, A., Verheijden, S., Van Veldhoven, P.P., Just, W., Devos, R., & Baes, M. (2012) J. Neuroinflammation, 9, 61.
20) Bottelbergs, A., Verheijden, S., Hulshagen, L., Gutmann, D.H., Goebbels, S., Nave, K.-A., Kassmann, C., & Baes, M. (2010) Glia, 58, 1532–1543.
21) Kassmann, C.M., Lappe-Siefke, C., Baes, M., Brügger, B., Mildner, A., Werner, H.B., Natt, O., Michaelis, T., Prinz, M., Frahm, J., & Nave, K.-A. (2007) Nat. Genet., 39, 969–975.
22) Kassmann, C.M., Quinters, S., Rietdorf, J., Möbius, W., Sereda, M.W., Nientiedt, T., Saher, G., Baes, M., & Nave, K.-A. (2011) FEBS Lett., 585, 2205–2211.
23) Igarashi, M., Schaumburg, H.H., Powers, J., Kishimoto, Y., Kolodny, E., & Suzuki, K. (1976) J. Neurochem., 26, 851–860.
24) Kobayashi, T., Shinnoh, N., Kondo, A., & Yamada, T. (1997) Biochem. Biophys. Res. Commun., 232, 631–636.
25) Lu, J.-F., Lawler, A.M., Watkins, P.A., Powers, J.M., Moser, A.B., Moser, H.W., & Smith, K.D. (1997) Proc. Natl. Acad. Sci. USA, 94, 9366–9371.
26) Pujol, A., Hindelang, C., Callizot, N., Bartsch, U., Schachner, M., & Mandel, J.L. (2002) Hum. Mol. Genet., 11, 499–505.
27) Fourcade, S., López-Erauskin, J., Galino, J., Duval, C., Naudi, A., Jove, M., Kemp, S., Villarroya, F., Ferrer, I., Pamplona, R., Portero-Otin, M., & Pujol, A. (2008) Hum. Mol. Genet., 17, 1762–1773.
28) López-Erauskin, J., Galino, J., Ruiz, M., Cuezva, J.M., Fabregat, I., Cacabelos, D., Boada, J., Martínez, J., Ferrer, I., Pamplona, R., Villarroya, F., Portero-Otín, M., Fourcade, S., & Pujol, A. (2013) Hum. Mol. Genet., 22, 3296–3305.
29) Deon, M., Marchetti, D.P., Donida, B., Wajner, M., & Vargas, C. (2016) Cell. Mol. Neurobiol., 36, 497–512.
30) Fan, C.-Y., Pan, J., Chu, R., Lee, D., Kluckman, K.D., Usuda, N., Singh, I., Yeldandi, A.V., Rao, M.S., Maeda, N., & Reddy, J.K. (1996) J. Biol. Chem., 271, 24698–24710.
31) Baes, M., Huyghe, S., Carmeliet, P., Declercq, P.E., Collen, D., Mannaerts, G.P., & Van Veldhoven, P.P. (2000) J. Biol. Chem., 275, 16329–16336.
32) Huyghe, S., Schmalbruch, H., Hulshagen, L., Van Veldhoven, P., Baes, M., & Hartmann, D. (2006) Am. J. Pathol., 168, 1321–1334.
33) Kassmann, C.M., Quintes, S., Rietdorf, J., Möbious, W., Sereda, M.W., Nientiedt, T., Saher, G., Baes, M., & Nave, K.-A. (2011) FEBS Lett., 585, 2205–2211.
34) Holzman, E., Teichberg, S., Abrahams, S.J., Citkowitz, E., Crain, S.M., Kawai, N., & Peterson, E.R. (1973) J. Histochem. Cytochem., 21, 349–385.
35) Kassmann, C.M. (2014) Biochimie, 98, 111–118.
36) Ando, S., Tanaka, Y., Toyoda, Y., & Kon, K. (2003) Neurochem. Res., 28, 5–13.