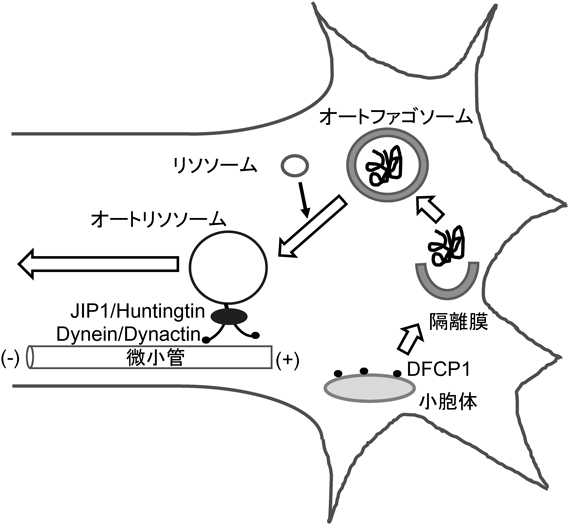
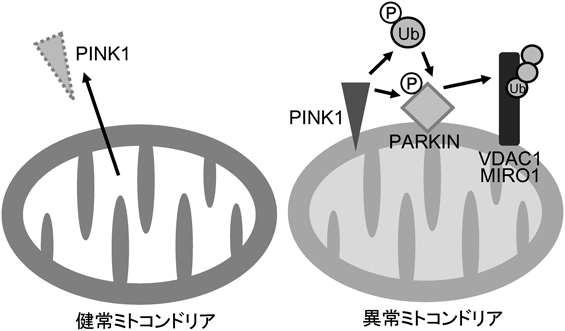
14) Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K., Kasai-Yoshida, E., Sawaura, N., Nishida, H., Hoshino, A., Ryujin, F., Yoshioka, S., Nishiyama, K., Kondo, Y., Tsurusaki, Y., Nakashima, M., Miyake, N., Arakawa, H., Kato, M., Mizushima, N., & Matsumoto, N. (2013) Nat. Genet., 45, 445–449, e1.
21) Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, J.P., Deng, H.X., Rahmani, Z., Krizus, A., McKenna-Yasek, D., Cayabyab, A., Gaston, S.M., Berger, R., Tanzi, R.E., Halperin, J.J., Herzfeldt, B., Van den Bergh, R., Hung, W.-Y., Bird, T., Deng, G., Mulder, D.W., Smyth, C., Laing, N.G., Soriano, E., Pericak-Vance, M.A., Haines, J., Rouleau, G.A., Gusella, J.S., Horvitz, H.R., & Brown, R.H. (1993) Nature, 362, 59–62.
23) Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., Kinoshita, Y., Kamada, M., Nodera, H., Suzuki, H., Komure, O., Matsuura, S., Kobatake, K., Morimoto, N., Abe, K., Suzuki, N., Aoki, M., Kawata, A., Hirai, T., Kato, T., Ogasawara, K., Hirano, A., Takumi, T., Kusaka, H., Hagiwara, K., Kaji, R., & Kawakami, H. (2010) Nature, 465, 223–226.
24) Cirulli, E.T., Lasseigne, B.N., Petrovski, S., Sapp, P.C., Dion, P.A., Leblond, C.S., Couthouis, J., Lu, Y.F., Wang, Q., Krueger, B.J., Ren, Z., Keebler, J., Han, Y., Levy, S.E., Boone, B.E., Wimbish, J.R., Waite, L.L., Jones, A.L., Carulli, J.P., Day-Williams, A.G., Staropoli, J.F., Xin, W.W., Chesi, A., Raphael, A.R., McKenna-Yasek, D., Cady, J., Vianney de Jong, J.M., Kenna, K.P., Smith, B.N., Topp, S., Miller, J., Gkazi, A., Al-Chalabi, A., van den Berg, L.H., Veldink, J., Silani, V., Ticozzi, N., Shaw, C.E., Baloh, R.H., Appel, S., Simpson, E., Lagier-Tourenne, C., Pulst, S.M., Gibson, S., Trojanowski, J.Q., Elman, L., McCluskey, L., Grossman, M., Shneider, N.A., Chung, W.K., Ravits, J.M., Glass, J.D., Sims, K.B., Van Deerlin, V.M., Maniatis, T., Hayes, S.D., Ordureau, A., Swarup, S., Landers, J., Baas, F., Allen, A.S., Bedlack, R.S., Harper, J.W., Gitler, A.D., Rouleau, G.A., Brown, R., Harms, M.B., Cooper, G.M., Harris, T., Myers, R.M., Goldstein, D.B., & Consortium, F.S. (2015) Science, 347, 1436–1441.
30) Valente, E.M., Abou-Sleiman, P.M., Caputo, V., Muqit, M.M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A.R., Healy, D.G., Albanese, A., Nussbaum, R., González-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W.P., Latchman, D.S., Harvey, R.J., Dallapiccola, B., Auburger, G., & Wood, N.W. (2004) Science, 304, 1158–1160.
32) Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., Kimura, Y., Tsuchiya, H., Yoshihara, H., Hirokawa, T., Endo, T., Fon, E.A., Trempe, J.F., Saeki, Y., Tanaka, K., & Matsuda, N. (2014) Nature, 510, 162–166.
35) Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M.L., Pahwa, J.S., Moskvina, V., Dowzell, K., Williams, A., Jones, N., Thomas, C., Stretton, A., Morgan, A.R., Lovestone, S., Powell, J., Proitsi, P., Lupton, M.K., Brayne, C., Rubinsztein, D.C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown, K.S., Passmore, P.A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A.D., Love, S., Kehoe, P.G., Hardy, J., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F., Schürmann, B., Heun, R., van den Bussche, H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M., Frölich, L., Hampel, H., Hüll, M., Rujescu, D., Goate, A.M., Kauwe, J.S., Cruchaga, C., Nowotny, P., Morris, J.C., Mayo, K., Sleegers, K., Bettens, K., Engelborghs, S., De Deyn, P.P., Van Broeckhoven, C., Livingston, G., Bass, N.J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C.E., Tsolaki, M., Singleton, A.B., Guerreiro, R., Mühleisen, T.W., Nöthen, M.M., Moebus, S., Jöckel, K.H., Klopp, N., Wichmann, H.E., Carrasquillo, M.M., Pankratz, V.S., Younkin, S.G., Holmans, P.A., O’Donovan, M., Owen, M.J., & Williams, J. (2009) Nat. Genet., 41, 1088–1093.
37) Yang, Y., Hentati, A., Deng, H.X., Dabbagh, O., Sasaki, T., Hirano, M., Hung, W.Y., Ouahchi, K., Yan, J., Azim, A.C., Cole, N., Gascon, G., Yagmour, A., Ben-Hamida, M., Pericak-Vance, M., Hentati, F., & Siddique, T. (2001) Nat. Genet., 29, 160–165.
40) Farrer, M.J., Hulihan, M.M., Kachergus, J.M., Dächsel, J.C., Stoessl, A.J., Grantier, L.L., Calne, S., Calne, D.B., Lechevalier, B., Chapon, F., Tsuboi, Y., Yamada, T., Gutmann, L., Elibol, B., Bhatia, K.P., Wider, C., Vilariño-Güell, C., Ross, O.A., Brown, L.A., Castanedes-Casey, M., Dickson, D.W., & Wszolek, Z.K. (2009) Nat. Genet., 41, 163–165.
45) Sherrington, R., Rogaev, E.I., Liang, Y., Rogaeva, E.A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J.F., Bruni, A.C., Montesi, M.P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau, P., Polinsky, R.J., Wasco, W., Da Silva, H.A., Haines, J.L., Perkicak-Vance, M.A., Tanzi, R.E., Roses, A.D., Fraser, P.E., Rommens, J.M., & St George-Hyslop, P.H. (1995) Nature, 375, 754–760.
47) Ramirez, A., Heimbach, A., Gründemann, J., Stiller, B., Hampshire, D., Cid, L.P., Goebel, I., Mubaidin, A.F., Wriekat, A.L., Roeper, J., Al-Din, A., Hillmer, A.M., Karsak, M., Liss, B., Woods, C.G., Behrens, M.I., & Kubisch, C. (2006) Nat. Genet., 38, 1184–1191.