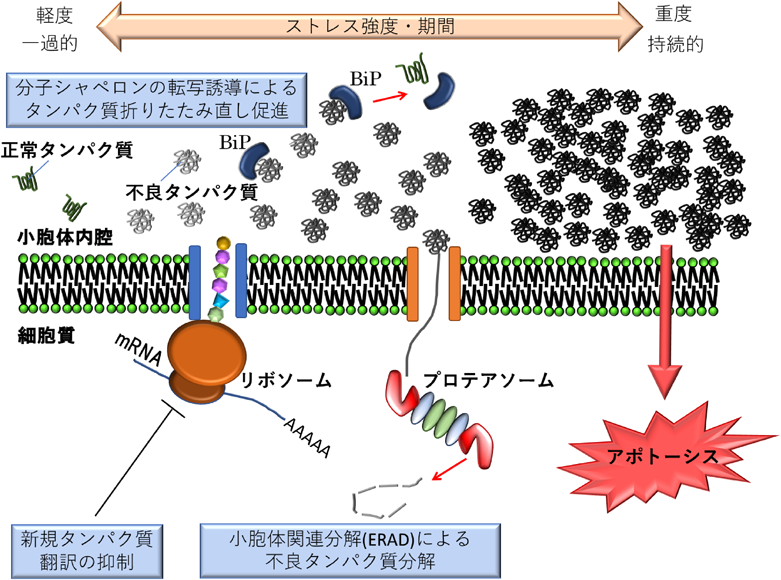
小胞体ストレスとは細胞がさまざまな内的あるいは外的環境変化にさらされることで,小胞体内腔においてタンパク質が正常に折りたたまれなくなり不良タンパク質として蓄積していく状態をいう.小胞体ストレスが生じる要因には,栄養飢餓,細胞内カルシウム濃度の撹乱,低酸素,変異タンパク質の発現,ウイルス感染などが知られる.小胞体ストレス状態になると細胞は,恒常性維持のためにタンパク質折りたたみの処理能力(フォールディングキャパシティ)を上げるために脂質合成を活性化し小胞体を拡張する.不良タンパク質の蓄積が比較的軽微な段階では,不良タンパク質排除のための応答機構によって不良タンパク質が取り除かれる.この応答機構は大きく分けて以下の三つの反応で構成される.すなわち,1)新たなタンパク質生合成を抑制するために,タンパク質の翻訳をブロックする.2)小胞体分子シャペロンの転写誘導によって折りたたみ不全タンパク質を折りたたみ直すことで不良タンパク質を減らす.そして,3)不良タンパク質を小胞体内腔から引きずり出し,プロテアソームに受け渡すことで不良タンパク質を分解する小胞体関連分解(ER associated degradation:ERAD)と呼ばれる機構によって,ストレス状態を軽減する.これら一連の応答機構はUPR(unfolded protein response)もしくは小胞体ストレス応答(ER stress response)と呼ばれる.まずはストレス状態を緩和するための反応によって,細胞を定常状態へ回復し保護しようと試みるのである.しかし,ストレス状態が重度であったり,長期間持続したりするような状況になると,UPRで対処しきれなくなる.その場合,細胞はアポトーシスを引き起こすことで,周囲の環境へ極力影響を及ぼさないようにする(図1).
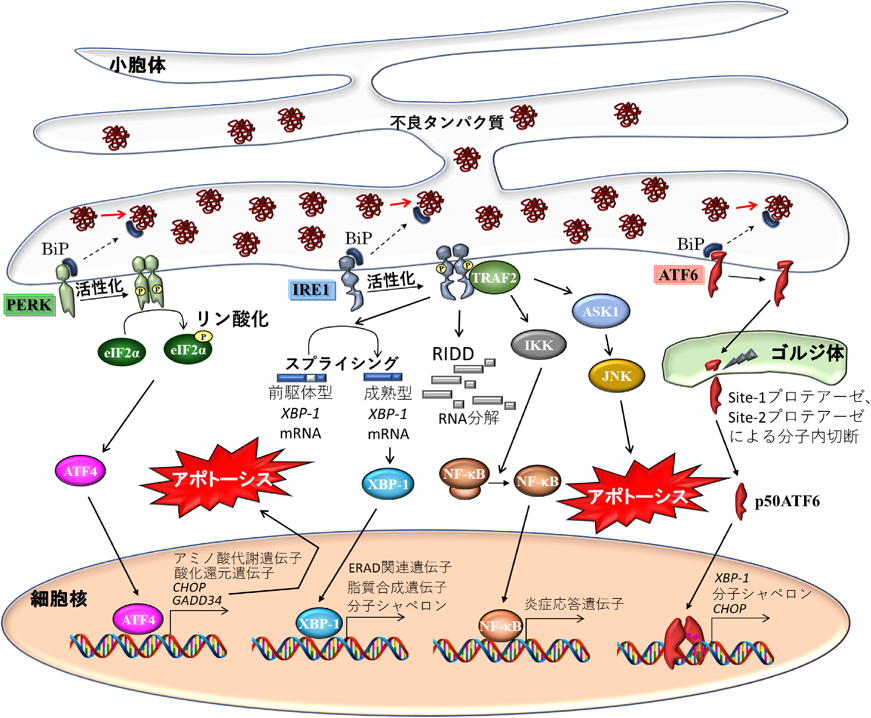
小胞体内腔の状態は,小胞体膜上に存在するストレスセンサーと呼ばれる1回膜貫通型タンパク質によってモニターされる.ストレスセンサーには,古典的ストレスセンサーと称されるユビキタスに発現する3種類のセンサータンパク質(IRE1, PERK, ATF6)と,組織特異的に発現するセンサータンパク質OASISファミリーが存在する.古典的ストレスセンサーは,定常状態では小胞体内腔側のドメインに小胞体分子シャペロンBiP/GRP78が結合しており,この結合によってセンサーは不活性状態を保っている.しかし,BiP/GRP78はセンサータンパク質よりも折りたたみ不全タンパク質の方に結合親和性があるため,小胞体ストレス状態によって小胞体内腔に不良タンパク質が蓄積すると,BiP/GRP78はセンサータンパク質から解離し,不良タンパク質と結合して,不良タンパク質の折りたたみ直しを媒介する.一方,BiP/GRP78が解離したセンサータンパク質は活性化し,UPR発動のシグナルを発信する(図2).これらセンサータンパク質の特徴について以下に述べる.
1)IRE1
IRE1(inositol requiring enzyme-1)は酵母から哺乳類に至るまで真核生物において広く保存されているセンサータンパク質であり,アミノ末端が小胞体内腔側を向いたI型膜タンパク質である.哺乳細胞においてはIRE1αとIRE1βの二つのパラログが存在し,IRE1αはほぼ全身に発現するのに対し,IRE1βは主に消化器官で発現している1).機能ドメインとして,細胞質側領域にキナーゼドメインとRNaseドメインを有する.小胞体ストレス下でBiP/GRP78が解離すると,二量体化あるいはオリゴマー化し,自己リン酸化により活性化する.活性化したIRE1はRNaseドメインで転写因子XBP-1のmRNAをスプライシングし,26 bpのRNAを切り出す.スプライシングされたXBP-1 mRNAによって翻訳されたタンパク質は機能的な転写因子XBP-1として働き,UPRに必要な標的分子の転写を誘導する.スプライシングされたXBP-1 mRNAをつなぐリガーゼは長らく不明であったが,近年複数のグループがほぼ同時期に,XBP-1 mRNAリガーゼとしてRtcBを同定した2–4).IRE1のRNaseドメインは,XBP-1のスプライシングだけでなく,RIDD(regulated IRE1-dependent decay)と呼ばれるシステムで特定のmRNAやmicroRNAを選択的に分解し,タンパク質の発現を抑制する他,アポトーシスに関わる機能も持つ.一方,活性化IRE1のキナーゼドメインは次のようにASK1およびIKKをリン酸化する.IRE1が活性化されるとアダプタータンパク質TRAF2がリクルートされる.このIRE1-TRAF2複合体にASK1あるいはIKKがリクルートされるとIRE1キナーゼドメインがそれらをリン酸化する.ASK1はリン酸化されると,続いてJNKをリン酸化し,JNK経路を活性化する.活性化したJNK経路はアポトーシスを引き起こす.一方,IKKのリン酸化はNF-κB経路の活性化を促し,炎症応答反応に関わる分子群の転写を誘導する.
2)PERK
PERK(PKR-like ER kinase)は細胞質側領域にキナーゼ活性を持つI型膜タンパク質である.BiP/GRP78が解離すると二量体あるいはオリゴマーを形成し,自己リン酸化によって活性化する.活性化したPERKは,翻訳開始因子のサブユニットeIF2αをリン酸化することで新たなタンパク質の翻訳を抑制する.eIF2αのリン酸化は一方で,uORF(upstream open reading frame)を持つ一部のmRNAの翻訳を促す.uORFを持つ転写因子ATF4はこのシステムにより翻訳誘導され,続いてアミノ酸代謝関連遺伝子や酸化還元分子の転写誘導が起こる.小胞体ストレスが持続すると,ATF4の下流でCHOPやGADD34の転写誘導が起こる.GADD34はprotein phosphatase 1と複合体を形成し,リン酸化eIF2αの脱リン酸化を促し,タンパク質翻訳シャットアウトから回復させる機能を持つ.ATF4により発現誘導されたCHOP, GADD34は小胞体ストレス依存的なアポトーシスを引き起こすことが知られている.
3)ATF6
ATF6(activating transcription factor 6)はアミノ末端が細胞質側に配向したII型膜タンパク質である.ATF6は脊椎動物においてATF6αとATF6βの2種類が存在する.遺伝子改変マウスを用いた解析からそれぞれ補完的に働くと考えられている.小胞体ストレス状態になると,BiP/GRP78が解離し,それによってATF6は小胞体からゴルジ体へと移行する.移行したゴルジ体においてsite-1プロテアーゼおよびsite-2プロテアーゼによって膜貫通領域が2段階に切断され(regulated intramembrane proteolysis:RIP),アミノ末端が膜から遊離する.遊離したアミノ末端はさらに核へ輸送され,転写因子として機能する.ATF6の標的遺伝子は,小胞体分子シャペロンBiP/GRP78やGRP94などが知られる.XBP-1およびCHOPもATF6によって転写誘導されることが報告されている.ホモ二量体を形成して転写因子として働くが,XBP-1とヘテロ二量体も形成する.
4)OASISファミリー(CREB3ファミリー)
OASISファミリーはATF6と構造がよく似たII型膜タンパク質であり,CREB3ファミリーとも呼ばれる.OASIS(別名CREB3L1),BBF2H7(別名CREB3L2),Luman(別名CREB3/LZIP),CREBH(別名CREB3L3),AIbZIP(別名CREB3L4/CREB4/TISP40)の5分子がある5–8).ATF6同様,RIPによってプロセシングを受け,アミノ末端側が転写因子として働く.OASISファミリーは組織特異的な発現パターンを示し,OASISは骨芽細胞やアストロサイト,大腸杯細胞などに高発現する.BBF2H7は軟骨細胞や神経細胞に優勢的に発現する.Lumanは樹状細胞,破骨細胞,神経細胞等に発現する.CREBHは肝細胞に発現し,AIbZIPは精巣や前立腺での発現が高い.
以上のストレスセンサーによって細胞は小胞体内腔の状態を常にモニタリングし,恒常性を保つが,ひとたびこの恒常性維持機構が破綻に向かうと,細胞はアポトーシスを起こし,種々の疾患発症へとつながっていく.次節からは小胞体ストレスが直接的あるいは間接的に関与する各種疾患を紹介する.
1)糖尿病
糖尿病は慢性的な高血糖状態を主徴とする疾患であり,膵臓β細胞から分泌されるホルモンであるインスリンの喪失あるいは作用不全で発症する.糖尿病には1型と2型があり,1型は膵臓β細胞の破壊・消失によってインスリンの産生・分泌ができなくなることで発症する.2型は,糖尿病になりやすい体質(遺伝的素因)と生活習慣による環境要因(肥満,運動不足など)が加わって,生体がインスリンの分泌低下やインスリンに対する抵抗性を獲得することで発症する.膵臓β細胞において小胞体ストレスが生じると,アポトーシスが引き起こされることでβ細胞の脱落が起こり,生体内でインスリン不足を来し糖尿病発症につながると考えられている.インスリンは未熟型のプレプロインスリンとして産生され,小胞体内でシグナルペプチド部分が切断されプロインスリンとなる.プロインスリンは続いてゴルジ体を経由して分泌顆粒に移行し,そこでさらに切断および分子内架橋を受けて成熟型のインスリンとなる.膵臓β細胞の機能障害と小胞体ストレスの関連性を顕著に示したのがAkitaマウスの例である9).Akitaマウスは2種類あるインスリン遺伝子のうちIns2遺伝子がコードするタンパク質の96番目のCysがTyrに変異しており,このC96Y変異によって分子内ジスルフィド結合ができず,ミスフォールドタンパク質として小胞体に蓄積し,小胞体ストレスを引き起こす.この小胞体ストレス惹起によって膵臓β細胞が喪失し糖尿病発症に至る.また,センサータンパク質PERKのノックアウトマウスが糖尿病様の表現型を呈することも知られている10).ヒトにおけるまれな疾患で,早期乳児期にインスリン依存型糖尿病症状を呈するWolcott-Rallison症候群は,PERK遺伝子に変異があることが明らかになっている11).小胞体ストレスによって発症する糖尿病の例としては他にも,Wolfram症候群があげられる.この疾患の原因遺伝子Wfs1に変異があると,膵臓β細胞の選択的な脱落を伴う若年発症の糖尿病となる12).2型糖尿病患者の膵臓組織由来のβ細胞では非糖尿病の膵組織と比べてUPR活性化マーカーが亢進する報告もあり13),小胞体ストレスが2型糖尿病発症にも関わる可能性が示唆されている.
2)神経変性疾患
神経変性疾患は,中枢神経系の特定の神経細胞群が障害を受け脱落することで神経機能を損なう病気である.代表的な神経変性疾患として,認知機能障害を引き起こすアルツハイマー病やクロイツフェルト・ヤコブ病,運動機能障害を呈するパーキンソン病,筋萎縮性側索硬化症(ALS)やハンチントン病があげられる.これら疾患の発症にも小胞体ストレスが関わっていることが知られている.疾患に共通する特徴として,変性タンパク質の蓄積が観察されることから,フォールディング病とも呼ばれる.たとえば,アルツハイマー病では細胞外にアミロイドβ,細胞内にタウタンパク質の蓄積が観察される.パーキンソン病においてはα-シヌクレイン,ALSでは変異スーパーオキシドジスムターゼ,ハンチントン病ではハンチンチンタンパク質の蓄積がみられる.プリオン病に分類されるクロイツフェルト・ヤコブ病では異常型プリオンが蓄積して発症する.このように,蓄積するタンパク質は疾患によって異なるが,変性タンパク質の蓄積で特定の神経細胞が障害され細胞死を起こすという共通性がみられる.神経変性疾患発症と小胞体ストレスとの関連性については,これまで多くの研究者によって精力的に研究され,優れた総説14, 15)もあるので,詳細はそちらを参照していただきたい.ここでは紙面の都合上,神経変性疾患と小胞体機能についての最近のトピックを紹介したい.
アルツハイマー病は大脳皮質ニューロンの脱落,大脳組織の萎縮による記憶障害・認知機能障害を引き起こす.特徴的な病理像として,アミロイドβタンパク質が細胞外に蓄積して形成される老人斑や,過剰にリン酸化した微小管結合タンパク質タウが細胞内部において繊維状に連結して変性した神経原線維変化がみられる.一方で,アルツハイマー病の病理所見としてあまり注目を受けていないが,コレステロール代謝,リン脂質代謝や細胞内カルシウムホメオスタシスの異常,ミトコンドリアの機能不全が生じていることがわかっている16–19).これらは潜在的に小胞体ストレスを惹起する現象である.近年,小胞体とミトコンドリアが相互作用する場MAM(mitochondria-associated ER membranes)が脂質代謝やカルシウムホメオスタシスの維持,ミトコンドリア機能の保持に重要な役割を果たしていることがわかってきた20).アミロイドβは主にこのMAMにおいて産生されることが示されている21).また,アルツハイマー病発症のリスク因子ApoE4タンパク質は,ミトコンドリアと小胞体のコンタクトサイトを増加させ,さらにリン脂質やコレステロールエステル生合成といったMAMの機能を亢進することが報告されており,このMAMの機能異常がアルツハイマー病と関連していることが示唆されている22).さらに,アルツハイマー病だけでなく,パーキンソン病,ALSにおいてもMAMの機能異常が報告されている23, 24).これら神経変性疾患の発症とMAM機能の関連性について,今後の詳細な解析を期待したい.
3)代謝性疾患(肥満,脂質異常症),循環器疾患
代謝性疾患に分類される肥満や脂質異常症の発症にも小胞体ストレスが関わることが指摘されている.野生型マウスに高脂肪食を与えると肝臓および脂肪組織において小胞体ストレスが惹起される.肥満状態ではPERKのリン酸化,eIF2αのリン酸化およびJNK活性が亢進する25).ヒトの肥満患者の皮下組織ではATF6シグナルにより小胞体ストレスマーカーBiP/GRP78の発現が亢進し,PERK経路のeIF2αのリン酸化も増強する26).マウスモデルを用いた研究において,脂質代謝の制御や脂肪性肝炎の進行にIRE1, PERK, ATF6の経路が関与することも報告されている27, 28).これら代謝性疾患では慢性炎症が生じており,この慢性炎症を引き起こすのが小胞体ストレスと考えられている.実際,野生型マウスに高脂肪食を与えると,脂肪組織において小胞体ストレスマーカーBiP/GRP78やATF4, CHOPなどの発現上昇と同時に,炎症応答マーカーであるIL-6やTNFαなどの発現上昇が誘導され,脂肪組織中にはマクロファージの浸潤が観察される29).この高脂肪食摂取による炎症応答は,小胞体ストレスを軽減するケミカルシャペロン4-PBA(4-phenylbutyric acid)やTUDCA(tauroursodeoxycholic acid)の投与により有意に抑制されることから,慢性炎症が小胞体ストレスによって惹起されることがわかる.動脈硬化や心筋梗塞などの循環器疾患も慢性炎症が主要な原因と考えられており,これら疾患においても小胞体ストレスが生じていることが報告されている30–32).
4)がん
がんは,現在,わが国の死因1位を占める疾患となっている.がんの特徴は1)自律的な増殖能,2)浸潤と転移能,3)悪液質(どんなに栄養摂取してもがん組織によって栄養が奪われ栄養失調状態になること)である.がん細胞は低酸素および低栄養環境下でも生存,増殖することができる.この低酸素・低栄養環境はまさに小胞体ストレスを惹起する環境であり,がん発症にUPRが関与していてもおかしくない.実際,複数種のがん細胞で小胞体分子シャペロンBiP/GRP78の発現亢進が認められ,がん細胞の増殖や転移に関わっている33).がん細胞は低栄養環境を克服するために細胞の近傍に新たに血管を新生することで栄養を確保しようとするが,この血管新生にもUPRが巧みに使われる.血管新生のキー分子である血管内皮増殖因子(VEGF)は,低酸素条件では転写因子HIF1によって発現誘導を受けることがよく知られるが,小胞体ストレス環境下でPERK-ATF4経路でも誘導される34).PERKによるシグナル経路は,血管新生だけでなくがん細胞の生存や転移にも働く.低グルコース条件下で活性化したPERKは,Aktシグナルを活性化し,続いてヘキソキナーゼIIのミトコンドリアへの移行を介してがん細胞の生存,増殖を促す35).また,PERKシグナルおよびそれによって誘導されたATF4は,上皮間葉転換(上皮細胞が遊走能,浸潤能を獲得し間葉系様の細胞に形質転換すること.epithelial-mesenchymal transition:EMTとも呼ばれる)と転移を促進する36, 37).IRE1αシグナルも,特定のがん細胞種において細胞増殖を促すことが報告されている.これは,IRE1αの下流のXBP-1を介した細胞周期因子CyclinA1の発現亢進による38).XBP-1は乳がん細胞や肝細胞がんにおいても発現が亢進しており,XBP-1の発現がBiP/GRP78の発現を誘導することでがん細胞の生存に寄与していると考えられている39, 40).ATF6はPERKシグナルおよびIRE1αシグナルと同様,VEGFの発現制御を介して血管新生に関わる報告があるものの41, 42),がん細胞の増殖や転移にどの程度寄与しているのかはあまりよくわかっていない.ただし,休眠期のがん細胞においては,Rheb-mTORシグナルを介してその生存に重要な働きをしている43).
5)高血圧
高血圧とは,血圧が正常範囲を超えて高く維持されている状態(ヒトにおいては,最高血圧が140 mmHg以上,あるいは,最低血圧が90 mmHg以上)と定義され,高血圧を放置しておくと脳梗塞や心筋梗塞,慢性腎臓病,糖尿病腎症などさまざまな合併症を引き起こす.内皮細胞の機能障害によって高血圧発症につながるが,その際,内皮細胞において血管拡張物質である一酸化窒素(NO)の合成低下や内皮依存性血管拡張反応(endothelium-dependent dilation:EED)の減弱を伴うことが知られる.小胞体ストレスと高血圧発症の関連性は,主に動物モデルを用いた研究により,その証拠が集められてきた.血圧上昇作用を有するアンジオテンシンII(Ang II)投与により作製された高血圧モデルマウスにおいて,Ang IIは小胞体ストレスマーカーとなるATF4やCHOP mRNAの発現誘導およびeIF2αのリン酸化を亢進する.一方で,このモデルマウスではNOの合成酵素であるeNOSのリン酸化の減弱や大動脈におけるEED低下が観察される44).このモデルにおいてケミカルシャペロン4-PBAやTUDCAを投与すると最高血圧の低下と,eNOSのリン酸化亢進およびEEDの増大が観察される44–46).このことからAng IIのシグナル下流ではUPRが発動し,内皮細胞による血圧の制御に関わることがうかがえる.他に次のような研究によっても高血圧症に小胞体ストレスが関与することが示されている.①正常血圧のSprague-Dawleyラットに小胞体ストレス誘導剤ツニカマイシン(Tm)を皮下投与すると,最高血圧が上昇し,大動脈平滑筋の線維化と細胞死を引き起こす45).②C57BL/6マウスへTmを投与すると最高血圧および最低血圧が上昇する46).③高血圧自然発生ラット(spontaneous hypertensive rat)に対して小胞体ストレスを抑制すると血圧の低下および大動脈の内皮細胞依存性収縮が弱まる47, 48).一方で,ヒトの研究においては,①肥満患者由来の内皮細胞では健常者由来の細胞に比べてUPR活性が亢進する49).②健常者由来の内皮細胞に脂質分子を添加するとUPRが活性化する50).③臨床試験においてTUDCAの経口投与が食事後高血糖に起因する内皮細胞機能障害を緩和することが報告されている51).これらの結果が示唆するのは,小胞体ストレスが内皮細胞の機能障害に関与することである.しかしながら,ヒトにおいて小胞体ストレスが高血圧につながるという直接的な証拠はまだ得られていない.今後の研究によってヒトにおいても高血圧発症に際して小胞体ストレスが引き金となるのか明らかになってくると思われる.
6)腎障害
腎障害にも小胞体ストレスが関わっていることが報告されている.ヒトの膜性腎炎に類似した症状を示すpassive Heymann腎炎モデルラットはeIF2αのリン酸化亢進とタンパク質の翻訳抑制が観察される52).微小変化型ネフローゼ症候群モデルラットでは腎組織でBiP/GRP78の発現が増加する53).Cybulskyらのグループは,巣状分節状糸球体硬化症モデルマウスにおいても小胞体分子シャペロンBiP/GRP78やGRP94の発現亢進およびeIF2αのリン酸化が増強されることを示した.興味深いことに,彼らはin vitroの実験で糸球体上皮細胞が細胞外マトリックスと相互作用すると小胞体ストレスを低減することを示唆するデータを示している54).糸球体上皮細胞が本来の小胞体機能を発揮するのに細胞外マトリックスからのシグナルが必要なのかもしれない.
Tm投与による急性腎障害モデルにおいても小胞体ストレスが亢進しており,このモデルに対してケミカルシャペロン4-PBAを処置すると,症状が緩和する55).この4-PBAによる腎障害の緩和は,小胞体ストレスによって発現誘導するCHOPが媒介しており,CHOPノックアウトマウスでは,Tm投与をしても急性腎障害が起こらない55).他方,免疫抑制剤として使用されるシクロスポリンは副作用として腎機能障害を引き起こすことが知られるが,シクロスポリン投与によって急性腎障害となったヒトの腎組織では,尿細管上皮細胞でのBiP/GRP78の発現亢進およびアポトーシス細胞の増加がみられる56).これは,シクロスポリンが脂質代謝調節転写因子SREBP-2の発現を促し,尿細管上皮細胞での脂質分子の蓄積を引き起こすことで小胞体ストレスを惹起し,最終的に細胞死につながるためと考えられている.
7)骨格形成異常
骨格の形成にも小胞体ストレスは重要な働きをする.糖尿病の項でも紹介したWolcott-Rallison症候群は糖尿病の症状以外にも多発性骨端異形成や骨減少を呈する.この疾患はPERK遺伝子の変異が原因であることが明らかとなっているが,このヒト疾患の症状と一致するように,PERKノックアウトマウスでも,骨形成不全が生じる57).また,PERKシグナル下流で誘導されるATF4も骨形成に関わる58).X連鎖性精神遅滞症候群であるCoffin-Lowry症候群は,主症状の精神運動発達遅滞以外にも骨格異常を呈することが知られており,その原因遺伝子はRSK2である.ATF4はこのRSK2によってリン酸化されることが報告され,骨格形成異常にRSK2-ATF4シグナルが関与することが示された.実際にATF4のノックアウトマウスも骨形成不全を呈する59).さらに,OASISファミリーに属するOASISのノックアウトマウスも骨形成不全の表現型を示し60),ヒトの先天性骨形成不全症の原因遺伝子の一つとしてもOASIS(CREB3L1)が遺伝解析から同定された61).これら骨形成不全が生じるメカニズムはそれぞれ異なり,PERKノックアウトは骨基質タンパク質の輸送制御異常を原因とした骨組織形成の不全であり,ATF4欠損においては骨組織の成熟化に必要なオステオカルシンや骨シアロタンパク質の発現が誘導されないために起こると考えられている.OASISは転写因子として骨基質タンパク質I型コラーゲンの転写誘導を行うため,OASIS欠損により骨基質産生が低下することで骨形成不全となる.骨基質を産生する細胞は骨芽細胞であり,骨形成に際し多量の骨基質タンパク質を産生する必要がある.そのため,骨芽細胞におけるUPRシグナルの破綻は骨基質タンパク質の正常な折りたたみや細胞外輸送の障害を引き起こし,骨形成不全を招くと考えられる.
軟骨形成にも小胞体ストレスセンサーによる制御が関わっている.OASISファミリーの一つであるBBF2H7のノックアウトマウスは重度の軟骨形成不全を呈し,出生直後に死に至る62).BBF2H7の標的遺伝子は輸送小胞の構成タンパク質Sec23aであり,BBF2H7をノックアウトすると輸送小胞を介した軟骨基質の細胞外への輸送が行われず,軟骨形成が障害される.BBF2H7ノックアウトマウスから単離した軟骨細胞では小胞体の異常な拡張が観察される.ただし,ヒト研究においてBBF2H7が軟骨形成不全に関わるかはまだ明らかとなっていないため,今後の解析が待たれる.
ここで紹介したように,小胞体ストレスが関与して発症する疾患は糖尿病や神経変性疾患をはじめとして,多岐にわたることが多くの研究から明らかとなってきた.疾患の発症要因が小胞体ストレスのみに限定されるものではないにせよ,小胞体ストレス,すなわち異常タンパク質の蓄積が直接的あるいは間接的に疾患発症につながることは疑いのない事実である.
近年,小胞体と他のオルガネラ間の連携についても興味深い知見が得られている.たとえば,先にも記述したようにミトコンドリアと小胞体のコンタクトサイトMAMが,細胞の恒常性維持に重要な働きを持っていることがわかってきた.このようなオルガネラ間連携の破綻によっても,これまで明らかになっていなかった分子メカニズムによって疾患発症につながっている可能性もある.他のオルガネラとの関わりによって発症する場合も考えると,これからは小胞体のみを対象とするのではなく,オルガネラ間相互作用も含めた統合的な機能の理解が必要になってくるであろう.小胞体機能の全容解明にますます目が離せない.
引用文献References
1) Tsuru, A., Fujimoto, N., Takahashi, S., Saito, M., Nakamura, D., Iwano, M., Iwawaki, T., Kadokura, H., Ron, D., & Kohno, K. (2013) Proc. Natl. Acad. Sci. USA, 110, 2864–2869.
2) Lu, Y., Liang, F.X., & Wang, X. (2014) Mol. Cell, 55, 758–770.
3) Kosmaczewski, S.G., Edwards, T.J., Han, S.M., Eckwahl, M.J., Meyer, B.I., Peach, S., Hesselberth, J.R., Wolin, S.L., & Hammarlund, M. (2014) EMBO Rep., 15, 1278–1285.
4) Jurkin, J., Henkel, T., Nielsen, A.F., Minnich, M., Popow, J., Kaufmann, T., Heindl, K., Hoffmann, T., Busslinger, M., & Martinez, J. (2014) EMBO J., 33, 2922–2936.
5) Kondo, S., Murakami, T., Tatsumi, K., Ogata, M., Kanemoto, S., Otori, K., Iseki, K., Wanaka, A., & Imaizumi, K. (2005) Nat. Cell Biol., 7, 186–194.
6) Kondo, S., Saito, A., Hino, S., Murakami, T., Ogata, M., Kanemoto, S., Nara, S., Yamashita, A., Yoshinaga, K., Hara, H., & Imaizumi, K. (2007) Mol. Cell. Biol., 27, 1716–1729.
7) Asada, R., Kanemoto, S., Kondo, S., Saito, A., & Imaizumi, K. (2011) J. Biochem., 149, 507–518.
8) Kanemoto, S., Kobayashi, Y., Yamashita, T., Miyamoto, T., Cui, M., Asada, R., Cui, X., Hino, K., Kaneko, M., Takai, T., Matsuhisa, K., Takahashi, N., & Imaizumi, K. (2015) J. Cell Sci., 128, 4353–4365.
9) Wang, J., Takeuchi, T., Tanaka, S., Kubo, S.K., Kayo, T., Lu, D., Takata, K., Koizumi, A., & Izumi, T. (1999) J. Clin. Invest., 103, 27–37.
10) Harding, H.P., Zeng, H., Zhang, Y., Jungries, R., Chung, P., Plesken, H., Sabatini, D.D., & Ron, D. (2001) Mol. Cell, 7, 1153–1163.
11) Delépine, M., Nicolino, M., Barrett, T., Golamaully, M., Lathrop, G.M., & Julier, C. (2000) Nat. Genet., 25, 406–409.
12) Inoue, H., Tanizawa, Y., Wasson, J., Behn, P., Kalidas, K., Bernal-Mizrachi, E., Mueckler, M., Marshall, H., Donis-Keller, H., Crock, P., Rogers, D., Mikuni, M., Kumashiro, H., Higashi, K., Sobue, G., Oka, Y., & Permutt, M.A. (1998) Nat. Genet., 20, 143–148.
13) Laybutt, D.R., Preston, A.M., Akerfeldt, M.C., Kench, J.G., Busch, A.K., Biankin, A.V., & Biden, T.J. (2007) Diabetologia, 50, 752–763.
14) Hetz, C. & Saxena, S. (2017) Nat. Rev. Neurol., 13, 477–491.
15) Remondelli, P. & Renna, M. (2017) Front. Mol. Neurosci., 10, 187.
16) Stefani, M. & Liguri, G. (2009) Curr. Alzheimer Res., 6, 15–29.
17) Pettegrew, J.W., Panchalingam, K., Hamilton, R.L., & McClure, R.J. (2001) Neurochem. Res., 26, 771–782.
18) Bezprozvanny, I. & Mattson, M.P. (2008) Trends Neurosci., 31, 454–463.
19) Wang, X., Su, B., Zheng, L., Perry, G., Smith, M.A., & Zhu, X. (2009) J. Neurochem., 109(Suppl 1), 153–159.
20) Szymański, J., Janikiewicz, J., Michalska, B., Patalas-Krawczyk, P., Perrone, M., Ziółkowski, W., Duszyński, J., Pinton, P., Dobrzyń, A., & Więckowski, M.R. (2017) Int. J. Mol. Sci., 18, E1576.
21) Schreiner, B., Hedskog, L., Wiehager, B., & Ankarcrona, M. (2015) J. Alzheimers Dis., 43, 369–374.
22) Tambini, M.D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., Sulzer, D., Area-Gomez, E., & Schon, E.A. (2016) EMBO Rep., 17, 27–36.
23) Rodríguez-Arribas, M., Yakhine-Diop, S.M., Pedro, J.M., Gómez-Suaga, P., Gómez-Sánchez, R., Martínez-Chacón, G., Fuentes, J.M., González-Polo, R.A., & Niso-Santano, M. (2017) Mol. Neurobiol., 54, 6287–6303.
24) De Mario, A., Quintana-Cabrera, R., Martinvalet, D., & Giacomello, M. (2017) Biochem. Biophys. Res. Commun., 483, 1096–1109.
25) Ozcan, U., Cao, Q., Yilmaz, E., Lee, A.H., Iwakoshi, N.N., Ozdelen, E., Tuncman, G., Görgün, C., Glimcher, L.H., & Hotamisligil, G.S. (2004) Science, 306, 457–461.
26) Sharma, N.K., Das, S.K., Mondal, A.K., Hackney, O.G., Chu, W.S., Kern, P.A., Rasouli, N., Spencer, H.J., Yao-Borengasser, A., & Elbein, S.C. (2008) J. Clin. Endocrinol. Metab., 93, 4532–4541.
27) Rutkowski, D.T., Wu, J., Back, S.H., Callaghan, M.U., Ferris, S.P., Iqbal, J., Clark, R., Miao, H., Hassler, J.R., Fornek, J., Katze, M.G., Hussain, M.M., Song, B., Swathirajan, J., Wang, J., Yau, G.D., & Kaufman, R.J. (2008) Dev. Cell, 15, 829–840.
28) Zhang, Q., Li, Y., Liang, T., Lu, X., Zhang, C., Liu, X., Jiang, X., Martin, R.C., Cheng, M., & Cai, L. (2015) Int. J. Biol. Sci., 11, 559–568.
29) Kawasaki, N., Asada, R., Saito, A., Kanemoto, S., & Imaizumi, K. (2012) Sci. Rep., 2, 799.
30) Hotamisligil, G.S. (2010) Cell, 140, 900–917.
31) Tabas, I. (2010) Circ. Res., 107, 839–850.
32) Azfer, A., Niu, J., Rogers, L.M., Adamski, F.M., & Kolattukudy, P.E. (2006) Am. J. Physiol. Heart Circ. Physiol., 291, H1411–H1420.
33) Miao, Y.R., Eckhardt, B.L., Cao, Y., Pasqualini, R., Argani, P., Arap, W., Ramsay, R.G., & Anderson, R.L. (2013) Clin. Cancer Res., 19, 2107–2116.
34) Pereira, E.R., Frudd, K., Awad, W., & Hendershot, L.M. (2014) J. Biol. Chem., 289, 3352–3364.
35) Hou, X., Liu, Y., Liu, H., Chen, X., Liu, M., Che, H., Guo, F., Wang, C., Zhang, D., Wu, J., Chen, X., Shen, C., Li, C., Peng, F., Bi, Y., Yang, Z., Yang, G., Ai, J., Gao, X., & Zhao, S. (2015) Sci. Rep., 5, 9065.
36) Pytel, D., Gao, Y., Mackiewicz, K., Katlinskaya, Y.V., Staschke, K.A., Paredes, M.C., Yoshida, A., Qie, S., Zhang, G., Chajewski, O.S., Wu, L., Majsterek, I., Herlyn, M., Fuchs, S.Y., & Diehl, J.A. (2016) PLoS Genet., 12, e1006518.
37) Hart, L.S., Cunningham, J.T., Datta, T., Dey, S., Tameire, F., Lehman, S.L., Qiu, B., Zhang, H., Cerniglia, G., Bi, M., Li, Y., Gao, Y., Liu, H., Li, C., Maity, A., Thomas-Tikhonenko, A., Perl, A.E., Koong, A., Fuchs, S.Y., Diehl, J.A., Mills, I.G., Ruggero, D., & Koumenis, C. (2012) J. Clin. Invest., 122, 4621–4634.
38) Thorpe, J.A. & Schwarze, S.R. (2010) Cell Stress Chaperones, 15, 497–508.
39) Fujimoto, T., Onda, M., Nagai, H., Nagahata, T., Ogawa, K., & Emi, M. (2003) Breast Cancer, 10, 301–306.
40) Shuda, M., Kondoh, N., Imazeki, N., Tanaka, K., Okada, T., Mori, K., Hada, A., Arai, M., Wakatsuki, T., Matsubara, O., Yamamoto, N., & Yamamoto, M. (2003) J. Hepatol., 38, 605–614.
41) Ghosh, R., Lipson, K.L., Sargent, K.E., Mercurio, A.M., Hunt, J.S., Ron, D., & Urano, F. (2010) PLoS One, 5, e9575.
42) Liu, L., Qi, X., Chen, Z., Shaw, L., Cai, J., Smith, L.H., Grant, M.B., & Boulton, M.E. (2013) Am. J. Pathol., 182, 1412–1424.
43) Schewe, D.M. & Aguirre-Ghiso, J.A. (2008) Proc. Natl. Acad. Sci. USA, 105, 10519–10524.
44) Kassan, M., Galán, M., Partyka, M., Saifudeen, Z., Henrion, D., Trebak, M., & Matrougui, K. (2012) Arterioscler. Thromb. Vasc. Biol., 32, 1652–1661.
45) Spitler, K.M. & Webb, R.C. (2014) Hypertension, 63, e40–e45.
46) Liang, B., Wang, S., Wang, Q., Zhang, W., Viollet, B., Zhu, Y., & Zou, M.H. (2013) Arterioscler. Thromb. Vasc. Biol., 33, 595–604.
47) Spitler, K.M., Matsumoto, T., & Webb, R.C. (2013) Am. J. Physiol. Heart Circ. Physiol., 305, H344–H353.
48) Choi, S.K., Lim, M., Byeon, S.H., & Lee, Y.H. (2016) Sci. Rep., 6, 31925.
49) Kaplon, R.E., Chung, E., Reese, L., Cox-York, K., Seals, D.R., & Gentile, C.L. (2013) J. Clin. Endocrinol. Metab., 98, E1505–E1509.
50) Tampakakis, E., Tabit, C.E., Holbrook, M., Linder, E.A., Berk, B.D., Frame, A.A., Bretón-Romero, R., Fetterman, J.L., Gokce, N., Vita, J.A., & Hamburg, N.M. (2016) J. Am. Heart Assoc., 5, e002574.
51) Walsh, L.K., Restaino, R.M., Neuringer, M., Manrique, C., & Padilla, J. (2016) Clin. Sci. (Lond.), 130, 1881–1888.
52) Cybulsky, A.V., Takano, T., Papillon, J., & Bijian, K. (2005) J. Biol. Chem., 280, 24396–24403.
53) Nakajo, A., Khoshnoodi, J., Takenaka, H., Hagiwara, E., Watanabe, T., Kawakami, H., Kurayama, R., Sekine, Y., Bessho, F., Takahashi, S., Swiatecka-Urban, A., Tryggvason, K., & Yan, K. (2007) J. Am. Soc. Nephrol., 18, 2554–2564.
54) Cybulsky, A.V., Takano, T., Papillon, J., Kitzler, T.M., & Bijian, K. (2011) Am. J. Physiol. Renal Physiol., 301, F496–F508.
55) Carlisle, R.E., Brimble, E., Werner, K.E., Cruz, G.L., Ask, K., Ingram, A.J., & Dickhout, J.G. (2014) PLoS One, 9, e84663.
56) Lhoták, S., Sood, S., Brimble, E., Carlisle, R.E., Colgan, S.M., Mazzetti, A., Dickhout, J.G., Ingram, A.J., & Austin, R.C. (2012) Am. J. Physiol. Renal Physiol., 303, F266–F278.
57) Wei, J., Sheng, X., Feng, D., McGrath, B., & Cavener, D.R. (2008) J. Cell. Physiol., 217, 693–707.
58) Saito, A., Ochiai, K., Kondo, S., Tsumagari, K., Murakami, T., Cavener, D.R., & Imaizumi, K. (2011) J. Biol. Chem., 286, 4809–4818.
59) Yang, X., Matsuda, K., Bialek, P., Jacquot, S., Masuoka, H.C., Schinke, T., Li, L., Brancorsini, S., Sassone-Corsi, P., Townes, T.M., Hanauer, A., & Karsenty, G. (2004) Cell, 117, 387–398.
60) Murakami, T., Saito, A., Hino, S., Kondo, S., Kanemoto, S., Chihara, K., Sekiya, H., Tsumagari, K., Ochiai, K., Yoshinaga, K., Saitoh, M., Nishimura, R., Yoneda, T., Kou, I., Furuichi, T., Ikegawa, S., Ikawa, M., Okabe, M., Wanaka, A., & Imaizumi, K. (2009) Nat. Cell Biol., 11, 1205–1211.
61) Symoens, S., Malfait, F., D’hondt, S., Callewaert, B., Dheedene, A., Steyaert, W., Bächinger, H.P., De Paepe, A., Kayserili, H., & Coucke, P.J. (2013) Orphanet J. Rare Dis., 8, 154.
62) Saito, A., Hino, S., Murakami, T., Kanemoto, S., Kondo, S., Saitoh, M., Nishimura, R., Yoneda, T., Furuichi, T., Ikegawa, S., Ikawa, M., Okabe, M., & Imaizumi, K. (2009) Nat. Cell Biol., 11, 1197–1204.
63) Moreno, J.A., Halliday, M., Molloy, C., Radford, H., Verity, N., Axten, J.M., Ortori, C.A., Willis, A.E., Fischer, P.M., Barrett, D.A., & Mallucci, G.R. (2013) Sci. Transl. Med., 5, 206ra138.
64) Celardo, I., Costa, A.C., Lehmann, S., Jones, C., Wood, N., Mencacci, N.E., Mallucci, G.R., Loh, S.H., & Martins, L.M. (2016) Cell Death Dis., 7, e2271.
65) Plate, L., Cooley, C.B., Chen, J.J., Paxman, R.J., Gallagher, C.M., Madoux, F., Genereux, J.C., Dobbs, W., Garza, D., Spicer, T.P., Scampavia, L., Brown, S.J., Rosen, H., Powers, E.T., Walter, P., Hodder, P., Wiseman, R.L., & Kelly, J.W. (2016) eLife, 5, e15550.
66) Ri, M., Tashiro, E., Oikawa, D., Shinjo, S., Tokuda, M., Yokouchi, Y., Narita, T., Masaki, A., Ito, A., Ding, J., Kusumoto, S., Ishida, T., Komatsu, H., Shiotsu, Y., Ueda, R., Iwawaki, T., Imoto, M., & Iida, S. (2012) Blood Cancer J., 2, e79.
67) He, L., Lee, J., Jang, J.H., Sakchaisri, K., Hwang, J., Cha-Molstad, H.J., Kim, K.A., Ryoo, I.J., Lee, H.G., Kim, S.O., Soung, N.K., Lee, K.S., Kwon, Y.T., Erikson, R.L., Ahn, J.S., & Kim, B.Y. (2013) Cell. Signal., 25, 552–560.
68) Zuleta, A., Vidal, R.L., Armentano, D., Parsons, G., & Hetz, C. (2012) Biochem. Biophys. Res. Commun., 420, 558–563.