神経細胞のAISは,シナプス電位を活動電位に変換する場として働く.これは,AISが構造と機能の面で,活動電位の発生に適した特性を持つことによる1–3).すなわち,AISはシナプス入力部である樹状突起・細胞体の近くにあるため,入力を効果的に受け取る(入力の減衰を最小限に抑える)ことができるのに加え,細く電気的にコンパクトなため,少ない入力でも脱分極することができる.さらに,AISには電位依存性Na(Nav)チャネルが高密度に集積する.これは,Navチャネルが足場タンパク質(ankyrinG)を介して細胞骨格(βIV spectrin, actin)や細胞接着分子(NrCAM, neurofascin186),細胞外基質(brevican, versican)に結合するためであり,このことによりAISでは活動電位の発生閾値が低くなり,小さな脱分極でも活動電位を発生することが可能になる.一方,AISにはNavチャネル以外にもさまざまなイオンチャネルが発現し,その種類や局在は個々の神経細胞ごとに異なることが知られている4).さらに,これらイオンチャネルは神経活動に応じてさまざまな制御を受ける.近年,AISの長さや軸索上での位置は神経細胞ごとに異なり,神経活動に応じて変化することが報告されている5).このことは,AISが神経活動生成の場であるだけでなく,その構造,イオンチャネルの活性や発現を可塑的に変化させることで,神経細胞の興奮性の精緻な調節に関わることを示している.最近の研究から,これらAISの構造とイオンチャネルの活性や発現は密接に連関していることがわかってきた.したがって,神経細胞における興奮性制御の仕組みを理解するには,これらAISの構造と機能の相互作用を明らかにすることが重要となる.本稿では,これまでに明らかとなってきたAISの構造と機能,その可塑性について概説し,さらにこれらが連関することで,どのように神経細胞の興奮性が効果的に調節されるのかについて述べる.
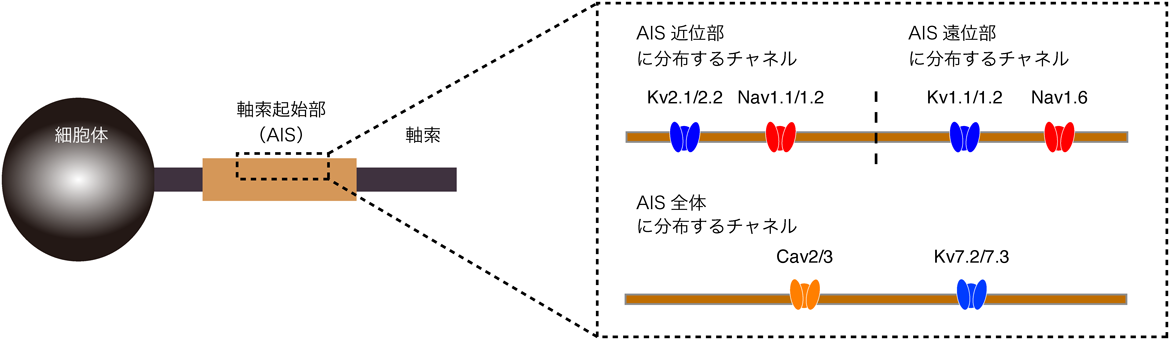
AISには,Navチャネルをはじめ,さまざまなイオンチャネルが発現する(図1).近年,これらイオンチャネルのサブタイプ,さらにそのAISでの分布や役割について多くのことがわかってきた.
AISに発現するNavチャネルとして,Nav1.1, Nav1.2, Nav1.6の三つのサブタイプが知られている.この中で,Nav1.6は活性化の閾値が低く,かつ脳のほぼすべての神経細胞種にみられることから,脳での活動電位発生を担う最も重要なチャネルといえる6–9).Nav1.6はAISの遠位部に分布する10, 11).この分布は,活動電位の発生効率を高める効果があり,活動電位の順行性伝播(軸索先端へ向かう興奮伝導)を増強する11–14).一方,Nav1.1とNav1.2は細胞種特異的に発現する7, 10, 11).すなわち,Nav1.1は抑制性の神経細胞に発現するのに対して,Nav1.2は興奮性の神経細胞に発現する.これらのチャネルはAISの近位部に位置するため,AISで発生した活電位が軸索から細胞体や樹状突起へと逆行性に伝播するのを助ける働きを持つ.
AISには,さまざまなサブタイプの電位依存性K+(Kv)チャネルが発現する7, 15).これらサブタイプの種類や発現量は細胞種ごとに異なり,このことが各神経細胞でみられる多様な発火様式に関わっている16).一般に,Kv1(Kv1.1と1.2)およびKv7(Kv7.2と7.3)は活性化の閾値が低く,静止膜電位付近でも活性化するため,短絡コンダクタンス(定常的なコンダクタンス)として,活動電位の発生を抑える働きを持つ17–20).加えて,Kv1は活動電位の時間短縮に関わり21, 22),Kv7は静止膜電位を維持することでNavチャネルの不活性化を防ぐ働きを持つことも知られている23).一方,Kv2(Kv2.1とKv2.2)は高い活性化の閾値を持つ.したがって,Kv2は通常,活動電位によって活性化し,活動電位の再分極相を加速することで,高頻度の発火を可能にする16).
AISには,電位依存性Ca2+(Cav)チャネルが発現し,活動電位の発生をさまざまな形で調節していることもわかってきた.たとえば,Cav2.3およびCav3.2は,活性化の閾値が比較的低く,活動電位に伴う後脱分極を増強することで,持続的な発火を引き起こす24).一方,Cav2.1およびCav2.2は活性化の閾値が高く,Ca2+依存性K+(BK)チャネルを活性化させることで,活動電位の再分極相を加速させる25).
このように,AISには複数のイオンチャネルが発現し,その種類や分布の違いによって興奮性の違いを生じることで,個々の神経細胞の出力様式が決定される.
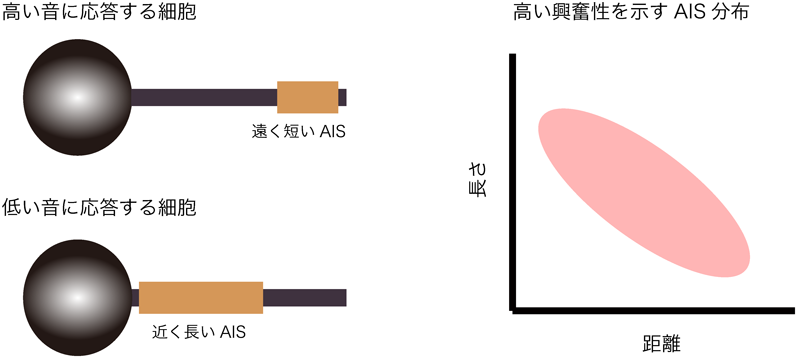
AISの位置や長さなどの構造特性は,細胞種ごとに異なり,神経細胞の興奮性に強く影響する26–29).AISの構造特性が神経細胞の興奮性へ及ぼす効果は,鳥類の層状核と呼ばれる聴覚神経核で詳しく調べられている27, 30).層状核は,哺乳類の内側上オリーブ核と相同の神経核であり,左右の耳に届く音の時間差を検出することで音源定位に関わる.層状核では,AISの長さと位置が,細胞が応答する音の周波数に応じて異なり,高い音に応答する細胞ほど短く,細胞体から遠くなる(図2).すなわち,AISの長さと細胞体からの距離の間には負の相関がある.この相関は神経細胞が興奮性を高める上で大きな意味を持つ.
AISを細胞体から離すことは二つの点で興奮性に影響する.一つは,樹状突起・細胞体コンパートメントが持つ大きな膜容量負荷の影響を小さくすることで,細胞の興奮性を高める.もう一つは,シナプス電流が細胞体からAISへと流入する際に,細胞体とAIS間の軸索部で失われる電荷量を増すことで,興奮性を低下させる.すなわち,AISが細胞体に近すぎる場合は,細胞体の膜容量負荷の影響を強く受けるために興奮性は低下し,一方,細胞体から遠すぎる場合も,AISに届く電荷が減るために興奮性は低下する.このように,細胞の興奮性は,AISが細胞体から適度な距離にある場合に最も高くなる.
AISの長さは,AISでのイオンチャネルの密度が一定の場合,AISでのイオンチャネルの総量と相関する.このことは,AISが長くなると,AISでのNa+とK+のコンダクタンスの両者が大きくなることを意味する.K+コンダクタンスは静止膜電位レベルでも活性化し,短絡コンダクタンスとして働く(上記参照).したがって,AISが長い場合には,AISが細胞体の近くにあるときに興奮性は高くなる.これは,AISが細胞体に近いことで細胞体から届く電荷が増えて,AISの短絡コンダクタンスに抗しやすくなることに加えて,AISが長いことでNa+コンダクタンスが大きくなり,樹状突起・細胞体による膜容量負荷に抗しやすくなることによる.一方,AISが短い場合には,AISが細胞体から離れているときに興奮性は高くなる.これは,AISが短いことで短絡コンダクタンスが小さくなり,少ない電荷でも脱分極できることに加えて,細胞体から離れていることで樹状突起・細胞体の膜容量負荷の影響が小さくなるためである.このように,AISの長さと距離は負の関係を持つことで,細胞の興奮性を高めている.この関係には,AISと樹状突起・細胞体でのコンダクタンス,すなわちイオンチャネルの発現が大きく影響する.樹状突起・細胞体のAISに対する影響は,マウス大脳皮質の錐体細胞でも報告されている31).
AISの構造やイオンチャネルはさまざまな時間スケールで変化し,このことにより神経細胞は状況に応じた出力調節を行うことができる.イオンチャネルの変化は,イオンチャネル型受容体の活性化による膜電位変化,もしくはAISでの代謝型受容体の活性化による制御を介するものであり,多くの場合,ミリ秒または秒単位の比較的速い変化となる.
1)イオンチャネル型受容体による制御
グルタミン酸作動性シナプスが,AISに形成されることはない.しかし,樹状突起・細胞体にグルタミン酸作動性シナプスからの大きな興奮性入力があると,興奮性入力は時間的に加重され,AISに持続的な脱分極を引き起こす.この脱分極は,AISでのNa+とK+のコンダクタンスのバランスを変えることで,神経細胞の興奮性を変化させる.たとえば,このシナプス電位加重による脱分極は,AISのKv1チャネルを不活性化させることで,AISでの活動電位を延長し,軸索終末からの伝達物質の放出を増強する21, 22).一方,このシナプス電位加重による脱分極は,AISのNavチャネルの不活性化も引き起こす.この脱分極によるNavチャネルの不活性化は細胞体に近い部位ほど強い.このため,マウス海馬の錐体細胞では大きな興奮性入力があると,一時的に活動電位の発生領域がAISの遠位部へ広がることが知られている32).これに対して,鳥類層状核の高い音に応答する細胞は,常に高頻度の入力にさらされているため,AIS自体を軸索の遠位部に配置する(上記参照)ことで,Navチャネルが不活性化することを避けている27).
マウス大脳皮質や海馬の錐体細胞では,AISにGABA作動性の軸索終末が軸索−軸索シナプスを形成し,活動電位の発生がAISへのGABA入力によって直接制御されることが知られている33).GABAA受容体は,リガンドであるGABAの結合によりCl−を通すイオンチャネルが開口する.一般に,成体脳では細胞内Cl−濃度が低いため,Cl−チャネルの開口はCl−を細胞内へ流入させることで過分極を起こす(つまり神経細胞の出力を減少させる方向に働く).興味深いことに,AISにはCl−を細胞外へ汲み出すカリウム−塩素共輸送体(KCC2)の発現が少なく,細胞内Cl−濃度が樹状突起・細胞体に比べて高い34, 35).このため,AISでのGABAA受容体の活性化はAISを脱分極させる34).このAISへのGABA入力が神経細胞の出力に及ぼす効果については議論が続いているが,現在のところ,AISでの短絡コンダクタンスを増すことで,神経細胞の出力を減少させるという考えが有力である36).
2)代謝型受容体による制御
AISでのイオンチャネルの活性は,代謝型受容体によっても制御されている.Navチャネルはセロトニン受容体(5HT1A)によって制御され,その効果は細胞種によって異なる37).マウス大脳皮質の錐体細胞では,5HT1A受容体はNavチャネルの活性化曲線を脱分極方向へシフトさせることで,AISでのNa+コンダクタンスを減少させる38).この調節効果は,Nav1.2に選択的であり,Nav1.6にはみられない.このため,5HT1A受容体の活性化は,活動電位の逆行性伝播を抑制するのに対して,順行性の興奮伝播にはほとんど影響しない.一方,スナネズミ内側上オリーブ核の神経細胞では,5HT1A受容体の活性化はAISでのNa+コンダクタンスを増加させることで,活動電位の発生を促進する39).これは,5HT1A受容体が過分極で活性化する陽イオンチャネル(HCNチャネル)を抑制し,過分極を生じることで,Navチャネルの不活性化を減少させるためである.
AISではCavチャネルも代謝型受容体による制御を受ける.マウス背側蝸牛神経核の抑制性神経細胞のAISでは,ドパミン受容体(D3)がプロテインキナーゼCを介して,Cav3.2を抑制する40).AISのCav3.2が抑制されることで,活動電位に伴う後脱分極は減弱するため,神経細胞の出力は減少する.一方,マウス海馬の顆粒細胞のAISでは,Cav3.2はムスカリン型アセチルコリン受容体(M1)によって活性化される41).Cav3.2の活性化は,AISでの細胞内Ca2+濃度を上昇させる.この細胞内Ca2+濃度上昇は,AISでのKv7の活性化曲線を過分極方向へシフトすることで,短絡コンダクタンスを減少させ,活動電位の発生を促進する.このように,AISではイオンチャネルの活性が細胞種特異的に調節され,このことが個々の神経細胞に応じた出力制御に関わっている.
AISの構造は神経活動に応じて変化する.この構造変化は恒常的可塑性として,神経回路の活動レベルを一定に保つ働きを持つ.AISの構造変化には,長さと位置の変化の2種類が知られており,いずれもイオンチャネルの変化に比べて遅く,多くの場合,数時間または数日の時間経過を要する.
1)位置の変化
AISの位置の変化は,ラット海馬の分散培養標本で最初に報告された.光感受性チャネル(チャネルロドプシン)発現下での光刺激,もしくは高K+液刺激によって神経細胞を長期間(2日間)脱分極させると,興奮性細胞ではAIS全体が遠位側へ移動し,細胞体から離れることで,興奮性が低下する42, 43).一方,マウス嗅球の分散培養標本では,長期間の脱分極は,興奮性細胞と抑制性細胞でAISに逆の変化を引き起こす44).すなわち,興奮性細胞ではAISが細胞体から離れる方向へ移動するのに対して,抑制性細胞では細胞体に近づく方向へ移動する.このことは,AIS可塑性が細胞種特異的に起こることを示している.これらAISの位置変化は,痙攣発作および脱髄のような神経細胞の過興奮性と関連する疾患モデルでもみられることから45, 46),神経回路に生じた過剰な活動を抑えるネガティブフィードバックの調節機構としての働きを持つと考えられている.
2)長さの変化
AISの長さの変化は,鳥類の大細胞核で最初に報告された47).大細胞核は哺乳類の蝸牛神経核と相同の神経核であり,聴神経からの入力を受けて層状核へと情報を送る.大細胞核では聴覚入力の消失に伴って,数日の時間経過でAISが伸長する.AISの伸長は,軸索遠位部のNa+コンダクタンスを増すことで,神経細胞の興奮性を高める.神経活動は,神経回路構築を維持する上で重要なことが知られている.したがって,この大細胞核の神経細胞で生じる興奮性の増加は,聴覚神経活動の消失を代償することで,中枢聴覚回路の維持に関わると考えられている.AISの伸長は,自閉症スペクトラム障害の一種であるAngelman症候群のマウスモデルでもみられる48).一方,AISの短縮は,脱髄疾患,脳卒中,外傷性脳損傷などでみられる45, 46, 49).また,AISの長さは,発達期の中枢感覚回路で求心性入力の出現に伴って短縮することが知られている50–52).このことは,AISの可塑性が,病的状況だけでなく,発達期の神経回路再編においても重要なことを示している.
6. AISの構造的可塑性に伴うイオンチャネルの変化
最近の研究から,AISの構造変化は,AISでのイオンチャネルの活性や発現の変化を伴うことがわかってきた.AIS構造による興奮性への効果には,AISに発現するイオンチャネルが強く影響するため,両者が同時に変化することは興奮性の調節において大きな意味を持つ.
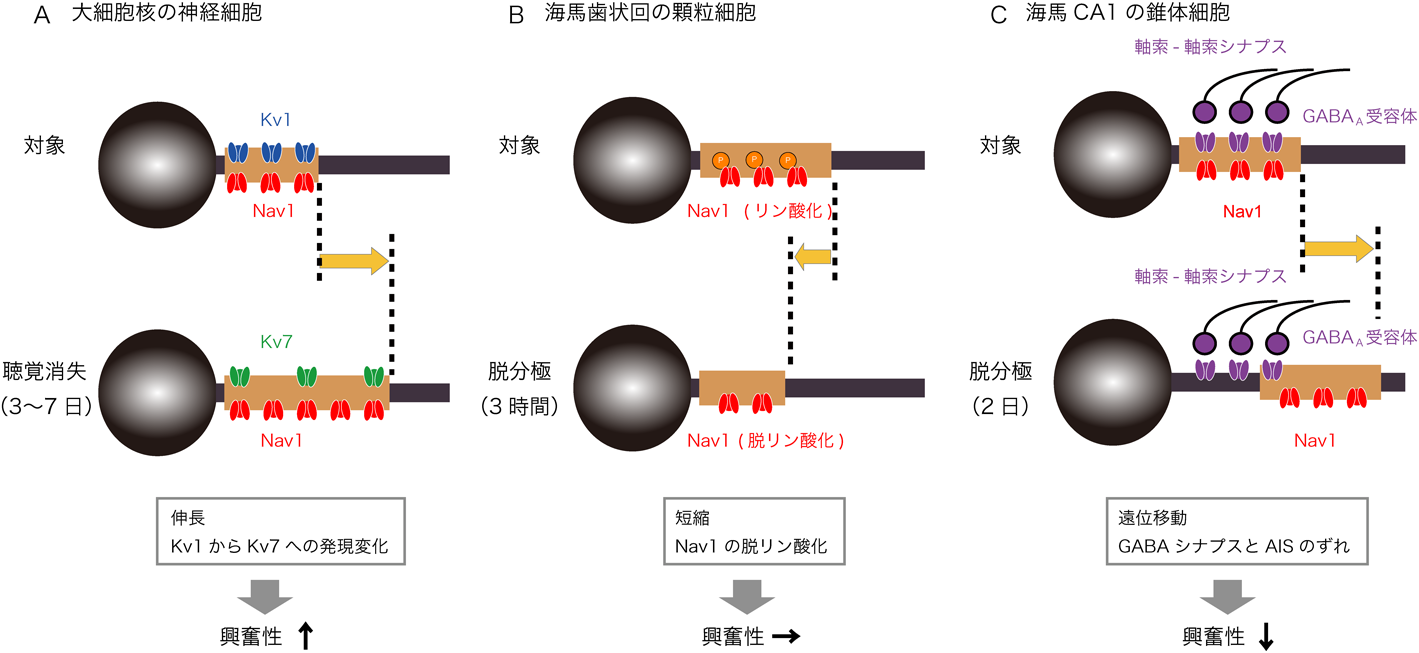
鳥類の大細胞核では,聴覚入力の消失によりAISが伸長する(上記参照).このAISの伸長に伴って,AISではKvチャネルの発現が,サブタイプ特異的に変化する.すなわち,Kv1.1は減少するのに対して,Kv7.2は増加する(図3A)53).Kv1.1は活性化の速度が速く,シナプス入力のような速い脱分極によっても強く活性化する.一方,Kv7.2は活性化の速度が遅いため,速い電位変化では開閉せず,定常的なコンダクタンスとして働く.したがって,Kv1.1とKv7.2の相補的な発現変化は,静止膜電位を大きく変えることなく,活動電位発生時の短絡コンダクタンスを減少させることになり,伸長したAISは効率的に聴神経活動の消失を代償することができる.このように,中枢の聴覚回路では,AISの構造と機能の変化が協調的に働くことで恒常性が維持される.
ラット海馬歯状回の顆粒細胞では,AISの構造と機能の変化は,拮抗的に働く(図3B)54).顆粒細胞を高K+液刺激により短期間(3時間)脱分極させると,AISは短縮し,同時にAISのNavチャネルが脱リン酸化される.AISの短縮は興奮性を低下させるのに対して,Navチャネルの脱リン酸化は興奮性を向上させる.したがって,両者は互いに打ち消し合い,興奮性は一定に維持される.しかしながら,現在のところ,この生理的意義はわかっていない.
ラット海馬CA1の錐体細胞では,長期間(2日)の脱分極により,AISが遠位方向へ移動し,細胞体から離れることで,興奮性が低下する(上記参照)43).この際,AISに発現するイオンチャネルはAISとともに移動する42).一方,GABA作動性の軸索−軸索シナプスの位置は移動せず,元の位置にとどまる(図3C)55).このため,軸索−軸索シナプスとAISとの間に位置のずれが生じ,このずれがAISの位置変化による興奮抑制の効果を増強する.これは,軸索−軸索シナプスが細胞体とAISの間にある軸索部の短絡コンダクタンスを増加させ,AISに届く電荷を減少させるためである.
AIS可塑性が生じる分子機構については,あまりわかっていない.AISの構造的可塑性は,Cav1チャネルを介した細胞内Ca2+濃度の変化に依存するが,下流のシグナル経路は細胞種や可塑性の種類によって異なる42–44).AISの構造変化に関わる分子として,カルシニューリンとサイクリン依存性キナーゼ5(cdk5)が知られている.カルシニューリンは,ラット海馬の興奮性細胞におけるAISの遠位方向への位置変化に関わり,cdk5は嗅球の抑制性細胞における近位方向への位置変化に関わる.これらの分子がAISの構造を再編する仕組みはまだわかっていないが,その仕組みにはAISタンパク質のパルミトイル化やリン酸化などの翻訳後修飾が関わる可能性が考えられている56).興味深いことに,カルシニューリンは,ラット海馬歯状回の顆粒細胞で,AISの短縮とNavチャネルの脱リン酸化を引き起こす54).また,cdk5はKv1のAISへの局在に関わる一方で57),ショウジョウバエのキノコ体ではAIS様構造を伸長させる58).このことは,AISの構造とイオンチャネルの変化には,共通のシグナル分子が関わることを示している.
AISでのイオンチャネルの変化はミリ秒から秒の単位で起こり,主に神経細胞の信号処理の調節に関わると考えられてきた.また,AISの構造変化ははるかに遅く,数日の単位で起こり,病的状況や発達期の神経回路の安定化や再編に関わると考えられてきた.しかしながら,最近の研究により,AISの構造変化はより短い時間スケール(数時間以内)でも起こり,長期増強や長期抑圧などの神経可塑性との相互作用を含め,神経細胞の信号処理に関わりうることがわかってきた.さらに,AISでのイオンチャネルの変化は,構造変化の効果を左右する.このことは,AISでの構造と機能の可塑性が,生理的な状況と病的な状況を問わず,互いに連関し,興奮性を幅広い時間スケールで制御することで,神経活動を巧妙に調節することを示している.
しかし,AISの可塑性については,多くの疑問が残されている.たとえば,なぜ脳領域や細胞種によって,AISの可塑性が異なるのか? 特に,個々の神経細胞でAIS可塑性を制御する仕組み,さらにその神経回路機能における役割を明らかにすることが必要である.また,AIS可塑性が時空間的にどのように制御され,他の神経可塑性とどのように相互作用するのかなどについても明らかにする必要がある.AISは神経細胞における出力決定の要であり,その異常はさまざまな精神神経疾患を引き起こすため59, 60),これらの問題を明らかにすることで,脳の生理機能や病態の理解が深まることが期待される.
謝辞Acknowledgments
本研究は科学研究費補助金基盤B, 新学術領域研究「スクラップ&ビルドによる脳機能の動的制御」,JSTさきがけ,上原記念生命科学財団の助成を受けて行われた.共同研究者である研究室メンバーに感謝いたします.
引用文献References
1) Debanne, D., Campanac, E., Bialowas, A., Carlier, E., & Alcaraz, G. (2011) Physiol. Rev., 91, 555–602.
2) Ogawa, Y. & Rasband, M.N. (2008) Curr. Opin. Neurobiol., 18, 307–313.
3) Kole, M.H.P. & Stuart, G.J. (2012) Neuron, 73, 235–247.
4) Grubb, M.S., Shu, Y., Kuba, H., Rasband, M.N., Wimmer, V.C., & Bender, K.J. (2011) J. Neurosci., 31, 16049–16055.
5) Kuba, H. (2012) J. Physiol., 590, 5571–5579.
6) Jenkins, S.M. & Bennett, V. (2001) J. Cell. Biol., 155, 739–746. Johnston, J., Forsythe, I.D., & Kopp-Scheinpflug, C. (2010) J. Physiol., 588, 3187–3200.
7) Lorincz, A. & Nusser, Z. (2008) J. Neurosci., 28, 14329–14340.
8) Colbert, C.M. & Pan, E. (2002) Nat. Neurosci., 5, 533–538.
9) Rush, A.M., Dib-Hajj, S.D., & Waxman, S.G. (2005) J. Physiol., 564, 803–815.
10) Van Wart, A., Trimmer, J.S., & Matthews, G. (2007) J. Comp. Neurol., 500, 339–352.
11) Hu, W., Tian, C., Li, T., Yang, M., Hou, H., & Shu, Y. (2009) Nat. Neurosci., 12, 996–1002.
12) Palmer, L.M. & Stuart, G.J. (2006) J. Neurosci., 26, 1854–1863.
13) Kole, M.H., Ilschner, S.U., Kampa, B.M., Williams, S.R., Ruben, P.C., & Stuart, G.J. (2008) Nat. Neurosci., 11, 178–186.
14) Baranauskas, G., David, Y., & Fleidervish, I.A. (2012) Proc. Natl. Acad. Sci. USA, 110, 4051–4056.
15) Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J.-Y., Cranstoun, S.D., Bennett, V., Scherer, S.S., & Cooper, E.C. (2006) J. Neurosci., 26, 2599–2613.
16) Johnston, J., Griffin, S.J., Baker, C., Skrzypiec, A., Chernova, T., & Forsythe, I.D. (2008) J. Physiol., 586, 3493–3509.
17) Dodson, P.D., Barker, M.C., & Forsythe, I.D. (2002) J. Neurosci., 22, 6953–6961.
18) Goldberg, E.M., Clark, B.D., Zagha, E., Nahmani, M., Erisir, A., & Rudy, B. (2008) Neuron, 58, 387–400.
19) Shah, M.M., Migliore, M., Valencia, I., Cooper, E.C., & Brown, D.A. (2008) Proc. Natl. Acad. Sci. USA, 105, 7869–7874.
20) Clark, B.D., Goldberg, E.M., & Rudy, B. (2010) Neuroscientist, 15, 651–668.
21) Kole, M.H., Letzkus, J.J., & Stuart, G.J. (2007) Neuron, 55, 633–647.
22) Shu, Y., Yu, Y., Yang, J., & McCormick, D.A. (2007) Proc. Natl. Acad. Sci. USA, 104, 11453–11458.
23) Battefeld, A., Tran, B.T., Gavrilis, J., Cooper, E.C., & Kole, M.H. (2014) J. Neurosci., 34, 3719–3732.
24) Bender, K.J. & Trussell, L.O. (2009) Neuron, 61, 259–271.
25) Yu, Y., Maureira, C., Liu, X., & McCormick, D. (2010) J. Neurosci., 30, 11858–11869.
26) Fried, S.I., Lasker, A.C., Desai, N.J., Eddington, D.K., & Rizzo, J.F. 3rd. (2009) J. Neurophysiol., 101, 1972–1987.
27) Kuba, H., Ishii, T.M., & Ohmori, H. (2006) Nature, 444, 1069–1072.
28) Kuba, H. & Ohmori, H. (2009) J. Physiol., 587, 87–100.
29) Kress, G.J., Dowling, M.J., Eisenman, L.N., & Mennerick, S. (2010) Hippocampus, 20, 558–571.
30) Adachi, R., Yamada, R., & Kuba, H. (2015) Neuroscientist, 21, 255–265.
31) Gulledge, A.T. & Bravo, J.J. (2016) eNeuro, 3, pii: ENEURO.0085-15.2016.
32) Scott, R.S., Henneberger, C., Padmashri, R., Anders, S., Jensen, T.P., & Rusakov, D.A. (2014) Nat. Commun., 5, 3817.
33) Howard, A., Tamas, G., & Soltesz, I. (2005) Trends Neurosci., 28, 310–316.
34) Szabadics, J., Varga, C., Molnar, G., Olah, S., Barzo, P., & Tamas, G. (2006) Science, 311, 233–235.
35) Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., & Kaila, K. (2008) J. Neurosci., 28, 4635–4639.
36) Woodruff, A.R., McGarry, L.M., Vogels, T.P., Inan, M., Anderson, S.A., & Yuste, R. (2011) J. Neurosci., 31, 17872–17886.
37) Cotel, F., Exley, R., Cragg, S.J., & Perrier, J.F. (2013) Proc. Natl. Acad. Sci. USA, 110, 4774–4779.
38) Yin, L., Rasch, M.J., He, Q., Wu, S., Dou, F., & Shu, Y. (2015) Cereb. Cortex, 27, 509–521.
39) Ko, K.W., Rasband, M.N., Meseguer, V., Kramer, R.H., & Golding, N.L. (2016) Nat. Neurosci., 19, 826–834.
40) Bender, K.J., Ford, C.P., & Trussell, L.O. (2010) Neuron, 68, 500–511.
41) Martinello, K., Huang, Z., Lujan, R., Tran, B., Watanabe, M., Cooper, E.C., Brown, D.A., & Shah, M.M. (2015) Neuron, 85, 346–363.
42) Grubb, M.S. & Burrone, J. (2010) Nature, 465, 1070–1074.
43) Evans, M.D., Sammons, R.P., Lebron, S., Dumitrescu, A.S., Watkins, T.B.K., Uebele, V.N., Renger, J.J., & Grubb, M.S. (2013) J. Neurosci., 33, 6950–6963.
44) Chand, A.N., Galliano, E., Chesters, R.A., & Grubb, M.S. (2015) J. Neurosci., 35, 1573–1590.
45) Harty, R.C., Kim, T.H., Thomas, E.A., Cardamone, L., Jones, N.C., Petrou, S., & Wimmer, V.C. (2013) Epilepsy Res., 105, 272–279.
46) Hamada, M.S. & Kole, M.H. (2015) J. Neurosci., 35, 7272–7286.
47) Kuba, H., Oichi, Y., & Ohmori, H. (2010) Nature, 465, 1075–1078.
48) Kaphzan, H., Buffington, S.A., Jung, J.I., Rasband, M.N., & Klann, E. (2011) J. Neurosci., 31, 17637–17648.
49) Baalman, K.L., Cotton, R.J., Rasband, S.N., & Rasband, M.N. (2013) J. Neurotrauma, 30, 741–751.
50) Cruz, D.A., Lovallo, E.M., Stockton, S., Rasband, M., & Lewis, D.A. (2009) J. Comp. Neurol., 514, 353–367.
51) Gutzmann, A., Ergül, N., Grossmann, R., Schultz, C., Wahle, P., & Engelhardt, M. (2014) Front. Neuroanat., 8, 11.
52) Kuba, H., Adachi, R., & Ohmori, H. (2014) J. Neurosci., 34, 3443–3453.
53) Kuba, H., Yamada, R., Ishiguro, G., & Adachi, R. (2015) Nat. Commun., 6, 8815.
54) Evans, M.D., Dumitrescu, A.S., Kruijssen, D.L., Taylor, S.E., & Grubb, M.S. (2015) Cell Reports, 13, 1233–1245.
55) Wefelmeyer, W., Cattaert, D., & Burrone, J. (2015) Proc. Natl. Acad. Sci. USA, 112, 9757–9762.
56) Yoshimura, T. & Rasband, M.N. (2014) Curr. Opin. Neurobiol., 27, 96–102.
57) Vacher, H., Yang, J.W., Cerda, O., Autillo-Touati, A., Dargent, B., & Trimmer, J.S. (2011) J. Cell Biol., 192, 813–824.
58) Trunova, S., Baek, B., & Giniger, E. (2011) J. Neurosci., 31, 10451–10462.
59) Wimmer, V.C., Reid, C.A., So, E.Y., Berkovic, S.F., & Petrou, S. (2010) J. Physiol., 588, 1829–1840.
60) Buffington, S.A. & Rasband, M.N. (2011) Eur. J. Neurosci., 34, 1609–1619.
著者紹介Author Profile
 久場 博司(くば ひろし)
久場 博司(くば ひろし)名古屋大学大学院医学系研究科教授.医学博士(京都大学).
略歴1997年九州大学医学部卒業後,九州大学医学部付属病院にて臨床研修.2003年京都大学大学院医学研究科博士課程修了,助教,講師,准教授を経て,11年より現職.
研究テーマと抱負音源定位に関わる聴覚神経回路の動作と形成の原理を研究する一方,近年は,その回路で見いだした新たな軸索可塑性のメカニズムや機能意義を明らかにすることを目指している.
ウェブサイトhttps://www.med.nagoya-u.ac.jp/medical_J/laboratory/basic-med/cell-science/physiol1/
趣味ジョギング,テニス.