記憶や学習は動物を動物たらしめる現象の一つであり,特に我々ヒトにおいては,社会生活を営む上で必須である.記憶や学習をつかさどる器官である脳は神経細胞やグリア細胞から成り立っているが,記憶・学習に直接関与しているのは神経細胞である.神経細胞は軸索と樹状突起からなる,高度に極性化された細胞である.軸索は別の神経細胞の樹状突起へと伸び,シナプスと呼ばれる接合部を介して信号を伝える.一つの神経細胞あたり数千以上ものシナプス結合が存在し,脳内では無数の神経ネットワークが形成される.このネットワークによる情報のやりとりが記憶,学習の基盤であると考えられている.
シナプスは情報を出力する側と受容する側とで構造的にも機能的にも異なっており,それぞれ,シナプス前部,シナプス後部と呼ばれる.神経細胞では,外部から入力された刺激は電気信号(活動電位)へと変換され,軸索終末であるシナプス前部へ伝えられ,神経伝達物質が分泌される(化学信号).神経伝達物質がシナプス後部膜にある神経伝達物質の受容体に結合することで,イオンチャンネルが開口し,化学信号は再び電気信号に変換され,次の細胞へ情報が伝えられる.シナプス後部膜にはシナプス後肥厚(post synaptic density:PSD)と呼ばれる特殊な膜領域が存在し,神経伝達物質の受容体やそれらをつなぎ止める足場タンパク質のような分子が高度に集積する.興味深いことに,PSDに集積する分子の多くが,パルミトイル化と呼ばれる脂質修飾を受けることが知られている1).
タンパク質の脂質修飾は,基質タンパク質の疎水性を高め,細胞膜やオルガネラ膜へと局在させる機能を有する.代表的な脂質修飾として,1)アシル化,2)プレニル化,3)グリコシルホスファチジルイノシトール(GPI)アンカー,4)コレステロール修飾の4種類が知られており,パルミトイル化はアシル化の一つである2).我々は,PSDにパルミトイル化修飾を受けるタンパク質が多く集積することに注目し,シナプスの機能がパルミトイル化によってどのように制御されうるのかに興味を抱き研究を行っている.本稿では,パルミトイル化修飾酵素について概観した後,パルミトイル化修飾を軸とした神経機能研究について,我々の研究成果を交えながら紹介する.また,最近同定した脱パルミトイル化酵素についても紹介するとともに,今後の可能性についても言及する.
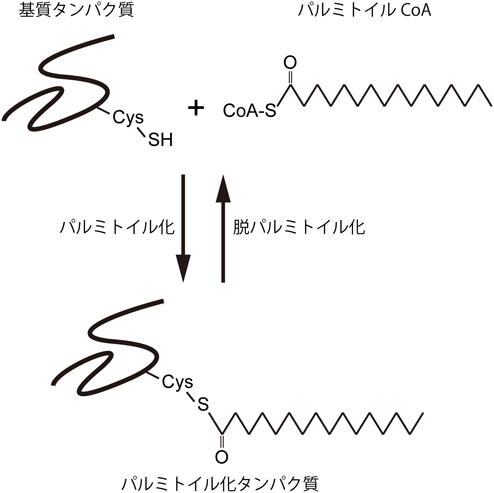
パルミトイル化は炭素数16の飽和脂肪酸であるパルミチン酸がタンパク質に共有結合する翻訳後修飾である1, 3).パルミチン酸がシステイン残基のチオール基に結合するS-パルミトイル化(図1)と,N末端に露出したシステイン残基のアミノ基に結合するN-パルミトイル化の2種類が存在する.しかし,これまでのところ,N-パルミトイル化を受けるタンパク質はHedgehogなど数種のタンパク質に限定されており4–6),比較的まれな修飾である.本稿ではより一般的なS-パルミトイル化について取り上げることとし,これ以降,パルミトイル化という表記はS-パルミトイル化として扱う.
パルミトイル化は,他の脂質修飾とは異なり,可逆的な修飾である(図1).すなわち,パルミチン酸転移酵素によるパルミトイル化反応と,脱パルミチン酸酵素による脱パルミトイル化反応が起きる.パルミトイル化と脱パルミトイル化のサイクルにより,基質タンパク質のパルミトイル化レベルや局在が規定されていると考えられている.
1979年,パルミトイル化を受けるタンパク質として,ウイルスの糖タンパク質が最初に報告された7, 8).その後,哺乳動物細胞をはじめ,幅広い生物種の多くのタンパク質のパルミトイル化が報告された.パルミトイル化は水溶性細胞質タンパク質,膜貫通型タンパク質の両方に起こりうる.パルミトイル化を受ける水溶性タンパク質の例として,H-RasやN-Rasなどの低分子量Gタンパク質,SNAP-25のようなsoluble N-ethylmaleimide sensitive factor attachment protein receptor(SNARE)などがあげられる1, 9).パルミトイル化を受ける膜貫通型タンパク質の例として,アドレナリン受容体のようなGタンパク質共役型受容体,α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid(AMPA)型グルタミン酸受容体(AMPA受容体)やN-methyl-D-aspartate(NMDA)型グルタミン酸受容体(NMDA受容体)などのイオンチャンネルなどがあげられる.代表的なパルミトイル化タンパク質を表1にまとめたので,参照されたい.
表1 代表的なパルミトイル化タンパク質| ファミリー | 主な例 |
|---|
| 三量体Gタンパク質(αサブユニット) | Gαs, Gαq, Gαoなど |
| 低分子量Gタンパク質 | H-Ras, N-Ras, Cdc42 isoform 2, RhoBなど |
| Srcファミリー | Fyn, Lyn, Yes, Lckなど |
| リン酸化酵素 | GRK6, CaMK1G, PI4K2Aなど |
| SNAREタンパク質 | SNAP-25, Syntaxin 1など |
| イオンチャンネル | GluA1, GluA2, GluN2A, GluN2B, GABAAγ2, Kv1.1, IP3R, CALHM1, HCNなど |
| Gタンパク質共役型受容体 | β2-アドレナリン受容体(β2AR),Rhodopsin, MC1R, S1P受容体など |
| 接着分子 | Integrin α6, CD4, CD8, CD9, NCAM140, Claudin-3, 4, 6, 7など |
| 足場タンパク質 | PSD-95, PSD-93α, PSD-93β, Ankyrin-Gなど |
| 各種酵素 | ABHD17, GAD65, BACE1, ELMOD2など |
| その他 | Huntingtin, Sortillin, Paralemmin, Calnexin, STING, CD151, δ-catenin, M6aなど |
3. パルミトイル化酵素ZDHHCファミリーと基質ペア
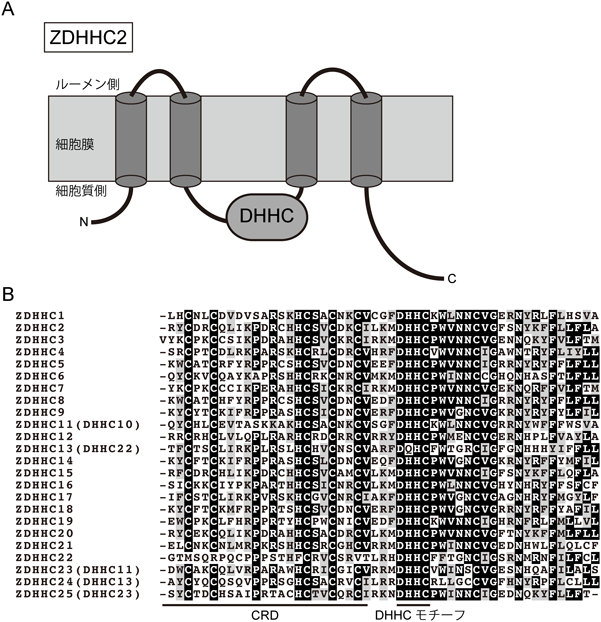
パルミトイル化タンパク質の同定以来,パルミトイル化を担う責任酵素の同定を目指した生化学的な研究が行われたが,長く責任酵素の単離には至らなかった.2000年前後に,酵母の順遺伝学的なスクリーニングにより,Erf2/Erf4複合体,およびAkr1がそれぞれ酵母のRas2とYck2をパルミトイル化する活性を持つことが示された10–12).Erf2とAkr1はそれぞれ4回または6回膜貫通領域を有し,ともにシステインリッチな領域(CRD)とアスパラギン酸–ヒスチジン–ヒスチジン–システイン(DHHC)モチーフからなる50アミノ酸程度の配列を,細胞質領域に有するタンパク質であった.変異導入実験の結果,DHHCモチーフがパルミチン酸転移活性に必須であることが示された.我々は,DHHC-CRDドメインの配列相同性を指標に,哺乳動物ゲノムからzinc finger DHHC(ZDHHC)22を除く23種類のZDHHCファミリータンパク質(図2A, B)を同定,クローニングし,その一部が実際にパルミトイル化反応を担うことを示した13).同時期に,他のグループからも哺乳動物のZDHHCタンパク質がパルミトイル化の責任酵素であるとの報告がなされ14, 15),ZDHHCファミリータンパク質が進化上保存されたパルミチン酸転移酵素であることが証明された.以降,酵母,哺乳動物以外にも,線虫,ショウジョウバエ,植物,原虫等においてZDHHCファミリーが見いだされている16–19).ごく最近,ヒトのZDHHC20とゼブラフィッシュのZDHHC15の結晶構造が報告され,ZDHHCタンパク質の反応機構が原子レベルで明らかにされた20).この報告で,ZDHHCタンパク質のCRDには2個のZn2+が結合すること,C末端の細胞質領域がDHHC-CRDドメインや膜貫通領域と相互作用し,活性に必須なドメインを形成することなどが明らかとなった.さらに,パルミトイル化阻害剤である2-bromopalmitate(2BP)との共結晶も報告され,DHHCモチーフのシステイン残基と2BPが共有結合している様子が原子レベルで解明された20).この結果は,DHHCモチーフのシステイン残基が活性中心であることを明確に示している20).また,2BPはZDHHCタンパク質の四つの膜貫通領域が形成する疎水性ポケットに入り込んでおり,供与基質であるパルミトイルCoAが反応の過程でこのポケットに挿入されうることも示された.
我々は,23種類の哺乳動物のZDHHCファミリー遺伝子パネルを用いた,網羅的な酵素–基質ペア同定法を報告した13, 21).この方法では,培養細胞に候補となる基質タンパク質と各ZDHHCタンパク質を共発現させ,[3H]パルミチン酸による代謝標識効率の増加を指標に,酵素–基質ペアを特定する.実際にこの方法を使って,我々はPSD-95がZDHHC2/3/7/15に,GαqがZDHHC3/7に,NeurochondrinがZDHHC1/3/7/11に,transmembrane AMPAR regulatory protein γ8(TARPγ8),Ca2+/calmodulin-dependent protein kinase IIα(CAMKIIα),Syd1がZDHHC3/7によってパルミトイル化されることを見いだした13, 22, 23).また,代謝標識法の代わりに後述するABE法による検出も行われており,δ-cateninがZDHHC5/20により24),MC1RがZDHHC13によりパルミトイル化されることが示された25).さらに,ZDHHCファミリー遺伝子の網羅的なノックダウンにより,ある基質タンパク質に対する責任酵素を同定する方法も考案されている.この方法により,ZDHHC6がZDHHC16によりパルミトイル化されることが示された26).さらにごく最近,HAP1細胞を用いて各ZDHHCのノックアウト細胞も構築され,酵素-基質ペアの同定に利用された27).ZDHHCファミリーの過剰発現系およびノックダウン系,さらにはパルミトイル化の検出法にはそれぞれ長所と短所があり,生理的な酵素–基質ペアの同定には,複数の実験手法による解析が必要である.これまで報告されている各ZDHHCファミリータンパク質の基質を表2にまとめたので,参照されたい.一方,これらの方法ではZDHHCタンパク質による基質認識機構を明らかにすることはできず,そのような研究は遅れていた.しかし最近,ZDHHC17のN末端に存在するアンキリンリピートとZDHHC17の基質であるSNAP25bのペプチドとの共結晶が報告され,ZDHHCタンパク質による基質認識機構が原子レベルで初めて明らかとなった28).他方,ZDHHCファミリータンパク質の多くはアンキリンリピートを有しておらず,それらのファミリーにおける基質認識機構についてはこれからの課題である.前述のZDHHC20の結晶構造をもとに,基質認識機構が詳細に明らかにされることが期待される.
表2 ZDHHCファミリーと基質ペア| ZDHHCファミリー | 主な基質 |
|---|
| ZDHHC1 | Neurochondrin |
| ZDHHC2 | PSD-95, SNAP-25, SNAP-23, eNOS, Fyn, CD151, CKAP4, ABCA1など |
| ZDHHC3 | PSD-95, SNAP-25, SNAP-23, Gαs, Gαq, Gαi2, GABAAγ2, eNOS, GluA1, GluA2, GAD65, Fyn, STREX, Neurochondrin, BACE1, NCAM140, TARPγ8, CAMKIIα, Syd1, CaMKIγなど |
| ZDHHC4 | BACE1 |
| ZDHHC5 | STREX, Flotillin-2, EZH2, δ-catenin |
| ZDHHC6 | Calnexin, IP3R |
| ZDHHC7 | PSD-95, SNAP-25, SNAP-23, GAP43, Gαs, Gαq, Gαi2, GABAAγ2, Fyn, STREX, Neurochondrin, BACE1, NCAM140, TARPγ8, CAMKIIα, Syd1, sortillin, PDE10A2など |
| ZDHHC8 | Cdc42 isoform 2, Paralemmin-1, GAD65, PSD-93, ABCA1, GRIP1 |
| ZDHHC9 | H-Ras, N-Ras, STREX, β2AR |
| ZDHHC11 (DHHC10) | Neurochondrin |
| ZDHHC12 | ABCA1 |
| ZDHHC13 (DHHC22) | Huntingtin, GAD65, SNAP-25, CTNND1, MC1R, Drp1 |
| ZDHHC14 | β2AR |
| ZDHHC15 | PSD-95, GAP43, Sortillin, CI-MPR, GABAAγ2, Fyn, BACE1, CD151など |
| ZDHHC16 | c-ABL, JAB1, DHHC6, Phospholamban |
| ZDHHC17 | Lck, SNAP-25, SNAP-23, Huntingtin, GAD65, STREXなど |
| ZDHHC18 | Lck, H-Ras, β2AR |
| ZDHHC19 | R-Ras, PDE10A2 |
| ZDHHC20 | Fyn, BACE1, ABCA1, EGFR, δ-catenin, IFITM3 |
| ZDHHC21 | Fyn, eNOS, Lck, ABCA1 |
| ZDHHC22 | KCNMA1 |
| ZDHHC23 (DHHC11) | NOS1, KCNMA1 |
| ZDHHC24 (DHHC13) | 未同定 |
| ZDHHC25 (DHHC23) | 未同定 |
各ZDHHCファミリーによるパルミトイル化修飾の生理的意義の解明を目指し,ZDHHCファミリー遺伝子のノックアウトマウスが作製され,解析されている.また,ZDHHC遺伝子の変異とヒト疾患との関連も報告されている.ZDHHCファミリー遺伝子のノックアウトマウスの表現型およびヒト疾患との関連を表3にまとめた29–46).
表3 ZDHHCファミリーのノックアウトマウスとヒト疾患との関連| ZDHHC ファミリー | ノックアウトマウス | ヒト疾患との関連 | 文献 |
|---|
| ZDHHC2 | — | 直腸がん,肝臓がん,肺がん | 29 |
| ZDHHC3 | オス特異的な低体重,GABAAγ2およびGAP-43のパルミトイル化レベルの減少 | — | 30 |
| ZDHHC5 | 胎生致死(50%),恐怖文脈条件づけの低下,Flotillin-2のパルミトイル化の減少 | — | 31, 32 |
| ZDHHC7 | 目立った表現型はなし | — | 30 |
| ZDHHC3/7 | 一部胎生致死,低体重,脳の低重量化 | — | 30 |
| ZDHHC8 | マッシュルーム型スパイン数の減少,興奮性シナプスの減少,統合失調症様の表現型,メス特異的なプレパルス抑制の低下,新しい環境における探索行動の低下 | 統合失調症 | 33, 34, 35 |
| ヘテロノックアウトマウス:作業記憶の低下 |
| ZDHHC9 | — | X連鎖知的障害 | 36, 37 |
| ZDHHC13 | R425X変異マウス:体重減少,寿命の短縮,肌のバリア機能低下,脱毛症,体毛のキューティクルの形成異常,骨粗しょう症 遺伝子トラップ:体重減少,脱毛症,進行性の神経失調,脳の重量低下,SNAP-25のパルミトイル化の減少 | ハンチントン舞踏病 | 38, 39, 40 |
| ZDHHC15 | — | X連鎖知的障害 | 41 |
| ZDHHC16 | 胎生致死(50%),眼の奇形(55%程度の胎児),心室壁の薄弱化と拡大化,心筋細胞核の異常形態化,心筋ミオパチー,除細動を含む心不全 | — | 42 |
| ZDHHC17 | 運動失調,認知機能低下,スパインの密度低下,興奮性シナプス伝達の低下,海馬における長期増強の低下,脳におけるSNAP-25, CSP, Synaptotagmin Iのパルミトイル化の低下 | ハンチントン舞踏病 | 43, 44 |
| ZDHHC13/17 | 胎生致死(胎生10日),胎盤の形成不全,Huntingtinのパルミトイル化減少 | — | 45 |
| ZDHHC21 | F233の欠失変異マウス:体毛の減少,毛包分化の遅延,毛包間表皮の過形成 | — | 46 |
遺伝子トラップ法により作製されたZDHHC5のノックアウトマウスは,メンデルの法則に従わず,予想の半分程度しか生まれてこない31).また,生まれてきたノックアウトマウスには恐怖文脈条件づけ記憶の有意な低下が認められる.これまでにZDHHC5の基質として同定されているδ-catenin(表2)は,ノックアウトマウスを使った研究により,ZDHHC5のノックアウトマウス同様,恐怖文脈条件づけ記憶の有意な低下が認められることが報告されている47).
ヒトにおけるZDHHC8のある種の多型が統合失調症のリスク因子として報告されている.さらに,ヒトZDHHC8遺伝子が存在する,22q11.2染色体領域の微小欠失が情緒障害や統合失調症の患者にしばしば認められ,このこともZDHHC8と統合失調症との関連性を示唆している.ZDHHC8のノックアウトマウスを用いた解析では,マッシュルーム型の成熟した樹状突起棘(スパイン)の密度が減少し33),メス特異的に,プレパルス抑制の低下,新しい環境における探索行動の低下など統合失調症様の表現型を示すことが報告されている34).ZDHHC8の基質は多数報告されているが33, 35)(表2),統合失調症との関連が明らかな基質は今のところ不明である.ZDHHC8はZDHHC5と同じサブファミリーに属すため,ZDHHC5による機能重複が考えられる.今後,ZDHHC8/5のダブルノックアウトマウスを用いた解析により,統合失調症と関連する基質が明らかにされることに期待がかかる.
男性に発症するX連鎖知的障害の関連遺伝子として,ZDHHC936, 37)およびZDHHC1541)が報告されている.しかし,本疾患と関連性が疑われるこれらZDHHCファミリーの基質タンパク質は同定されていない.ZDHHC9がH/N-Rasをパルミトイル化することと(表2),Rasファミリーがシナプス可塑性に重要な役割を果たすこと48)を考え合わせると,ZDHHC9の変異によりH/N-Rasのパルミトイル化レベルが低下することが本疾患の発症原因であるのかもしれない.ZDHHC9ノックアウトマウスの解析が待たれる.
N-ethyl-N-nitrosoureaによる変異スクリーニングにより,ZDHHC13のナンセンス変異(R425X)のマウスが見いだされた38).この変異により生じる短いZDHHC13はHuntingtin(表2)のパルミトイル化活性をほとんど失っていた.この変異マウスは,成長不良で,およそ半分のマウスが生後7.5か月で死亡する.一方,別のグループが報告した遺伝子トラップ法により作製されたZDHHC13のノックアウトマウスは,成長不良はみられるものの,生後12か月までの生存率は野生型と違いがみられなかった40).また,SNAP-25(表2)のパルミトイル化レベルが若干低下していたが,Huntingtinのパルミトイル化には影響を与えなかった.なぜ二つのノックアウトマウス系統が異なる表現型を示すのかは不明である.ZDHHC13はHuntingtin結合タンパク質として同定されていたZDHHC17とともにサブファミリーを形成する15).そのため,両者は互いに機能相補する可能性が考えられたため,ZDHHC13/17のダブルノックアウトマウスが作製された.しかし,ZDHHC13/17のダブルノックアウトマウスは胎生10.5日ごろに致死となった45).このマウス由来の胚性線維芽細胞による解析では,Huntingtinのパルミトイル化レベルが約25%低下していた45).このことは,ZDHHC13/17が生体内でHuntingtinを基質とすることを示唆している.
ZDHHC16のノックアウトマウスは新生マウスの11%しか生まれず,一部胎生致死となる42).55%のZDHHC16のノックアウトマウスには単眼もしくは両眼の欠損,低形成などの奇形がみられる.さらに,心室壁の薄弱化と拡大化,心筋細胞の核の異常形態化,心筋ミオパチー,除細動を含む心不全などの心臓の異常が多くみられることから,ZDHHC16は眼や心臓の発生に重要な役割を果たすと考えられる.PhospholambanがZDHHC16の基質タンパク質として同定されており,これらの表現型との関連性が示唆されている.
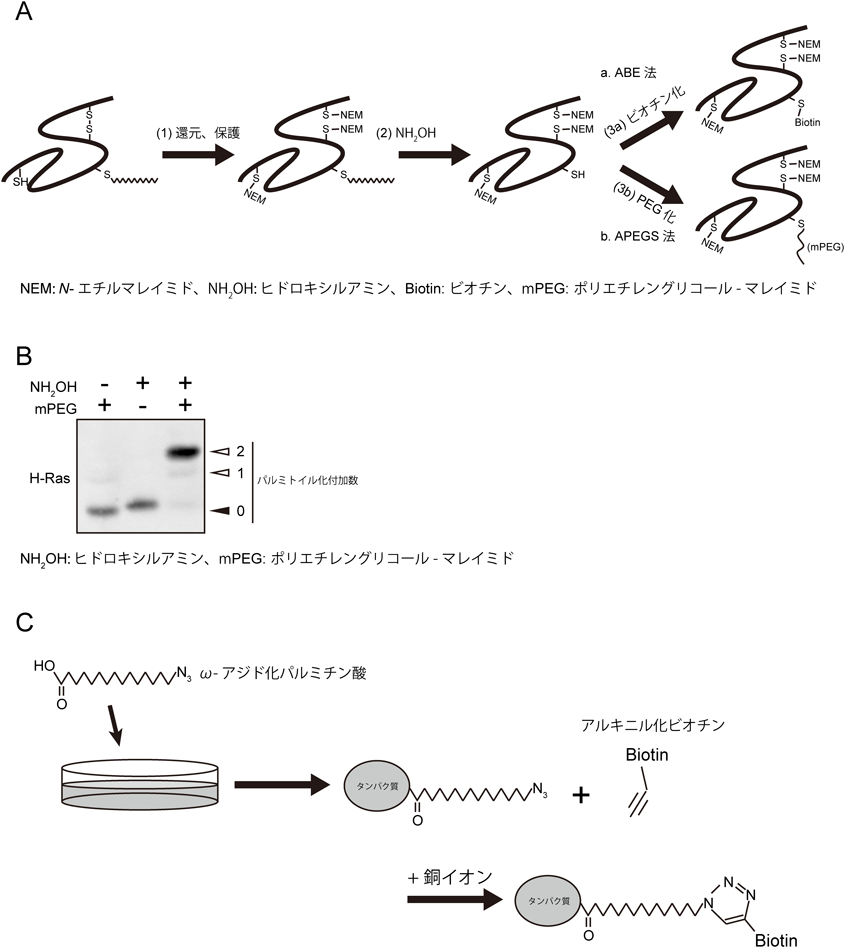
パルミトイル化脂質修飾の検出法としては,長い間放射性同位体で標識されたパルミチン酸([3H]あるいは[125I]パルミチン酸)を用いた代謝標識法が利用されていたが,その検出には多大な費用と時間が必要であった.2004年には,Greenらによってacyl-biotinyl exchange(ABE)法という,細胞や組織から効率よくパルミトイル化タンパク質を標識,精製する手法が開発された49).ABE法は,(1)タンパク質を還元処理した後,パルミトイル化されていないシステイン残基をN-エチルマレイミドにより保護し(アルキル化),(2)パルミトイル化システインのチオエステル結合を,ヒドロキシルアミンにより切断,脱パルミトイル化し,(3)新たにできたフリーのシステイン残基をビオチン化する,という三つの反応ステップから成り立っている[図3A-(3a)].ABE法を利用することで特定のタンパク質のパルミトイル化の有無を調べること,さらには質量分析と組み合わせることで,網羅的なパルミトイル化タンパク質の同定(パルミトーム解析)が可能となる.2006年にはABE法を利用した,酵母におけるパルミトーム解析が報告され,12種類の既知の基質に加え,35種類の新規のパルミトイル化タンパク質が同定された50).この報告は,ABE法がパルミトイル化研究において強力な研究手法となることを示しており,ラットのシナプトソーム画分,培養細胞,植物の根などの異なるサンプルにおけるパルミトームが次々と報告された51–53).以降,パルミトイル化タンパク質をより特異的かつ高感度に検出できるように,ABE法の改変手法が報告されている54).一方,代謝標識法やABE法では,あるパルミトイル化タンパク質におけるパルミトイル化量比(ストイキオメトリー)やパルミトイル化数(モノ,ジ,トリなど)についての情報を得ることができない.そこで,我々は,ABE法を改変した手法として,acyl-PEGyl exchange gel shift(APEGS)法を開発した55).APEGS法は,ABE法と同様の手順でフリーのシステイン残基の保護とパルミトイル化の脱離を行うが,ビオチン化する代わりに,ポリエチレングリコール–マレイミド(mPEG)をパルミトイル化されていたシステイン残基に結合させる[図3A-(3b)].これにより,パルミトイル化を電気泳動上の移動度の変化で検出できるようになる(図3B).したがって,1)非パルミトイル化(シフトなし)とパルミトイル化(シフトあり)の状態を区別して検出できるので,同一分子におけるパルミトイル化のストイキオメトリーを計算することが可能となる.また,2)パルミトイル化されていたシステイン残基の数に依存して,移動度が変動することから,同一分子に付加されうるパルミトイル化の個数を調べることができる.現時点では,APEGS法は同一分子におけるパルミトイル化のストイキオメトリーを調べることのできる唯一の方法である.APEGS法により,ラットの海馬由来初代培養神経細胞内のPSD-95の約90%が2か所のパルミトイル化修飾を受けることが示された55).また,H-RasもPSD-95同様,80%以上の分子が2か所のパルミトイル化修飾を受けることが示された55)(図3B).
また,放射性同位体の代わりに,bioorthogonal(生体内の反応を邪魔しない)なパルミチン酸アナログであるω-アジド化パルミチン酸とクリックケミストリーを利用した代謝標識法が開発されている56, 57).アジド基は銅イオン依存的にアルキニル基と特異的に反応することから,銅イオンとアルキニル化ビオチンを加えることで,パルミトイル化修飾をビオチン化することが可能となる(クリック反応,図3C).アルキニル化パルミチン酸とアジド化ビオチンの組合わせでのクリック反応も可能であり,こちらの組合わせの方が非特異的な反応が少ないとする報告もある58).また,アジド基はホスフィン基とも生理的な条件で特異的に反応する(Staudinger ligation).アジド基とホスフィン基の反応は銅イオンに依存しないので,より生理的な条件での標識が可能である.bioorthogonalなパルミチン酸を使った代謝標識法は,ABE法同様,網羅的なパルミトイル化タンパク質の同定に利用できる57).代謝標識法は,ABE法やAPEGS法とは異なり,パルスチェイス法と組み合わせ,パルミトイル化タンパク質のターンオーバー速度を算出することに適している59).
6. 神経細胞におけるパルミトイル化タンパク質の機能
PSD-95はシナプス後部膜のタンパク質の2%ほどを占める主要な足場タンパク質であり,三つのPSD-95/Dlg/ZO-1(PDZ)ドメインとSrc-homology 3(SH3)ドメイン,グアニル酸キナーゼ様ドメインからなる.PSD-95はN末端から3番目と5番目の二つのシステイン残基にパルミトイル化修飾を受け60),パルミトイル化依存的にPSDへと集積する61).PSD-95は三つのPDZドメインを介して,Neuroligin等のシナプス接着分子,SynGAP等のシグナル伝達分子,さらにはNMDA受容体のようなシナプス伝達に必須の分子と直接結合する.また,PSD-95はAMPA受容体とは直接結合しないものの,TARPと呼ばれる付属サブユニットを介してAMPA受容体をPSDにつなぎ止める62).PSD-95の過剰発現により,AMPA受容体のシナプス後膜での発現が増加し,AMPA受容体を介したシナプス伝達が増強する63).PSD-95の二つのパルミトイル化部位に変異を入れた,パルミトイル化欠損変異体の過剰発現ではこの効果はみられない.逆に,PSD-95のノックダウンあるいは,PSD-95, PSD-93のダブルノックアウトにより,AMPA受容体を介したシナプス伝達が有意に減少する64, 65).これらの結果は,PSD-95のPSDでの発現量が,AMPA受容体のPSDでの発現量を規定する重要な要因であることを示唆している.
最近,神経活動依存的にδ-cateninのパルミトイル化レベルが亢進することが示された24).δ-cateninのパルミトイル化酵素であるZDHHC5は,定常状態ではPSD-95やFynと結合する66).FynはZDHHC5の533番目のチロシンをリン酸化し,ZDHHC5のエンドサイトーシスを阻害する.神経活動が活性化することで,Fynの酵素活性およびPSD-95との結合親和性が低下し,ZDHHC5は効率的にエンドサイトーシスされ,樹状突起内のリサイクリングエンドソームへと輸送される.その結果,ZDHHC5と細胞質中のδ-cateninが空間的に近づき,ZDHHC5によるδ-cateninのパルミトイル化が亢進する.その後,ZDHHC5とパルミトイル化されたδ-cateninがともに細胞膜上へと輸送され,δ-cateninはカドヘリンを安定化させ,AMPA受容体をシナプス膜へとリクルートする66).前述のように,δ-cateninのノックアウトマウスは認知機能の低下と恐怖条件づけにおける記憶力低下を示すことが報告されており47),δ-cateninのパルミトイル化と神経機能の相関が個体レベルで示されている.
7. 古典的な脱パルミトイル化酵素と新規の脱パルミトイル化酵素ABHD17
これまで,脱パルミトイル化酵素としては,acyl-protein thioesterase 1(APT1),APT2, palmitoyl protein thioesterase 1(PPT1),PPT2が知られていた.APT1はGαiの脱パルミトイル化酵素として精製された67).その後,APT1はin vitroでH-Rasにも脱パルミトイル化酵素活性を示すこと,培養細胞への過剰発現によりGαsやeNOSの脱パルミトイル化を促進することが報告された68).初代培養神経細胞に対し,APT1のノックダウンや,特異的阻害剤(FD196やFD253)を作用させることにより,スパインの大きさが小さくなったことから,APT1はスパインの形成に関与すると考えられている69).
PPT1はH-Rasの脱パルミトイル化酵素として精製された70).後にPPT1はリソソームのタンパク質として報告がなされ71),細胞質側に存在するパルミトイル化タンパク質を直接脱パルミトイル化するとは考えられなかった.一方,その後の研究で,PPT1がシナプス前部に見いだされ72),シナプス小胞のプールを維持するために機能することも示唆された73).また,PPT1は乳児神経セロイドリポフスチン症の原因遺伝子として古くから報告されていた74).この病気は,神経細胞などのリソソームにセロイドと呼ばれる自家蛍光を発する不溶性の色素が蓄積する,リソソーム蓄積病の一種である.PPT1のノックアウトマウスでも同様に,セロイドがリソソーム内に認められ,ヒト疾患と類似した神経変性やてんかんなどの症状を示した75).おそらく,パルミトイル化タンパク質がオートファジー経路で分解される際,PPT1による脱パルミトイル化が必須であり,PPT1の機能異常によりリソソーム内にパルミトイル化タンパク質が蓄積し,セロイドを形成するのであろう.Sarkarらはチオエステル結合を切断できるヒドロキシルアミンの誘導体である,N-(tert-butyl)hydroxylamine(NtBuHA)が乳児神経セロイドリポフスチン症の治療薬の候補となりうると考えた76).NtBuHAは無毒であり,神経セロイドリポフスチン症の患者由来の初代培養神経細胞のリソソームに蓄積したセロイドを効果的に除去することができた.また,PPT1のノックアウトマウスにNtBuHAを与えることで,脳の重量増加,神経細胞の増加を促すことができ,脳萎縮の進行を遅らせることに成功している.この事実は,NtBuHAが血液脳関門を通り抜けて効果を発揮していることを示しており,チオエステラーゼを模した低分子化合物が神経セロイドリポフスチン症の治療薬になりうることを示唆している.PPT1の基質タンパク質を同定することを目的として,PPT1の結合分子の網羅的探索が行われ,同定された23分子のうち,CRMP1, DBH, MAP1Bの3分子がパルミトイル化タンパク質であった77).今後,これらの分子がセロイド形成や神経変性に関与するかどうか解析が待たれる.また,他のPPT1の基質パルミトイル化タンパク質の同定も今後の課題であろう.
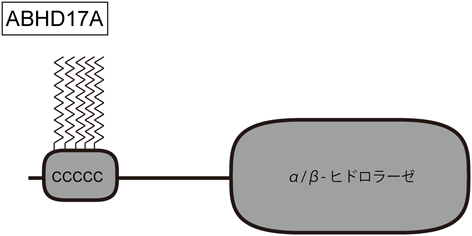
PSD-95はPSDで脱パルミトイル化される(後述)ので,細胞質タンパク質やリソソームタンパク質であるAPT1/PPT1のみがPSD-95の脱パルミトイル化酵素であるとは考えにくく,他の脱パルミトイル化酵素の存在が予想されていた.しかし,その正体は長く不明であった.Martinらはセリン加水分解酵素遺伝子群の中に,脱パルミトイル化を担う酵素が含まれると仮説を立て,脱パルミトイル化反応阻害剤としてhexadecylfluorophosphonate(HDFP)を合成した59).HDFP処理によりH-RasやPSD-95などの脱パルミトイル化が阻害されるとともに,21種類のセリン加水分解酵素の活性が阻害されたことから,これらの中に脱パルミトイル化酵素が存在することが示唆された.我々は,Martinらによって見いだされた21種類のセリン加水分解酵素の多くがα/β-ヒドロラーゼドメインを有することに着目し,HDFPに感受性を示したセリン加水分解酵素および,α/β-hydrolase domain-containing(ABHD)ファミリーを網羅的に単離し,PSD-95に対して脱パルミトイル化活性を示すかどうか調べた55).HEK293TおよびCOS7細胞にPSD-95と候補酵素を共発現させ,PSD-95の脱パルミトイル化が促進するかどうかを調べたところ,ABHD17A, B, Cのそれぞれの過剰発現によりPSD-95の脱パルミトイル化が大きく促進することが明らかとなった.ABHD17A, Bは自身にパルミトイル化修飾を受けるシステイン残基を5か所(図4),ABHD17Cは4か所有しており,ABHD17Bのパルミトイル化修飾システイン残基をすべてセリンに置換したパルミトイル化欠損ABHD17BはPSD-95に対する脱パルミトイル化活性を失った.ラット海馬初代培養神経細胞を用いたABHD17Aの局在解析の結果,ABHD17Aはパルミトイル化依存的に細胞膜,リサイクリングエンドソームおよびスパインへ局在した.ラット海馬初代培養神経細胞に対して,ABHD17A, B, Cの多重ノックダウンを行ったところ,PSD-95のパルミトイル化の半減期が約2倍に延長し,定常状態でのPSD-95のパルミトイル化が増加した.これらの結果から,ABHD17A, B, CがPSD-95の脱パルミトイル化酵素であることが示された.ほぼ同時期に,海外のグループもABHD17の過剰発現,およびノックダウンによる解析を行い,N-RasがABHD17により脱パルミトイル化されることを示した78).ラット海馬初代培養神経細胞を使った我々の解析では,ABHD17はH-RasなどのPSD-95以外の分子をほとんど基質としなかったことから55),少なくとも神経細胞においては,ABHD17ファミリーはPSD-95に対する特異性が高いと考えられる.最近,神経細胞におけるABHD17の基質として新たにmicrotubule-associated protein 6(MAP6)が報告された79).今後もABHD17の基質タンパク質が同定されていくと思われる.
8. パルミトイル化サイクルによるタンパク質の局在制御
脱パルミトイル化はパルミトイル化修飾の特徴的かつ重要な反応である.パルミトイル化と脱パルミトイル化のサイクルにより,基質タンパク質がダイナミックに局在を変化させることが可能となる.このようなパルミトイル化サイクルは,恒常的に生じる場合と,外部刺激により制御されうる場合があり,どちらの場合も基質タンパク質の局在や機能を制御する上で重要である.本節では,パルミトイル化サイクルによる基質タンパク質の局在変動に関する知見を紹介する.
脱パルミトイル化による局在変化が最初に報告された分子はGαsである.細胞をGタンパク質共役型受容体である,アドレナリン受容体のアゴニストで処理することで,Gαsの脱パルミトイル化が亢進し,Gαsが細胞質へと離散し,シグナル伝達効率が低下する80).その後,光退色後蛍光回復法(fluorescence recovery after photobleaching:FRAP)を用いた解析により,GαqとGαoはパルミトイル化サイクルによりゴルジ体と細胞膜の間を行き来していることが示された22, 81).また,H/N-Rasもパルミトイル化サイクルにより,恒常的に細胞膜とゴルジ体の間をシャトルしていることが報告された82, 83).
一方,神経シナプスの代表的な足場タンパク質であるPSD-95は恒常的にパルミトイル化と脱パルミトイル化を受けており,グルタミン酸による神経刺激により脱パルミトイル化が亢進することが見いだされた84).従来,PSD-95のような足場タンパク質はさまざまなタンパク質を細胞膜近傍に固定したり,膜貫通型タンパク質を安定につなぎ止めたりするものだと考えられていたため,PSD-95がダイナミックにパルミトイル化と脱パルミトイル化のサイクルを繰り返しているという報告は驚きであった.逆にいえば,PSD-95のパルミトイル化サイクルには機能的な意味が含まれていると考えられる.後述するように,我々は,PSD-95のパルミトイル化サイクルとシナプス構築やシナプス機能との関連を提唱している.
9. PSDナノドメインとPSD-95のパルミトイル化サイクル
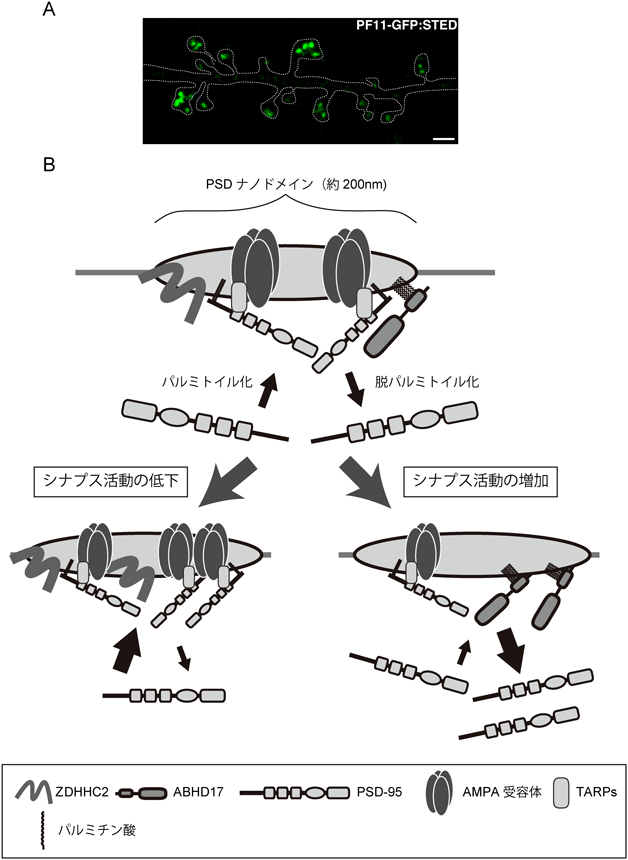
我々は内在性PSD-95のパルミトイル化状態を時空間的に可視化するための分子プローブを開発した.ファージディスプレイ法により,パルミトイル化PSD-95に対する単鎖組換え抗体の選別を行い,PF11と名づけた組換え抗体を得ることに成功した85).PF11はHEK293T細胞内,神経細胞内ともに,PSD-95を認識したが,パルミトイル化を受ける2か所のシステインをそれぞれセリンに置換したパルミトイル化欠損変異体を認識せず,パルミトイル化PSD-95特異的に結合すると考えられた.PF11のcDNAにGFPを融合させ,海馬初代培養神経細胞に発現させたところ,内在性のパルミトイル化PSD-95を可視化することができた.さらに,stimulated emission depletion(STED)超解像顕微鏡により観察を行ったところ,PSDがこれまで知られていなかったさらに小さなナノメートルサイズの構造(PSDナノドメイン)から構成されていることが明らかとなった85).PSDナノドメインは一つあたり200 nm程度の大きさで,一つのスパインあたり,1~4個程度含まれていた(図5A).重要なことに,PSDの大きさとPSDナノドメインの数には強い相関関係がみられ,PSDの大きさはPSDナノドメインの数で規定される可能性が示唆された.PSDナノドメインにはAMPA受容体が含まれており,シナプス伝達を担う最小の構造単位である可能性を示唆している(図5B上).さらに,PSD-95-GFPおよびPF11-GFPを用いたFRAP解析の結果,光退色後1時間で蛍光回復した割合は,PSD-95-GFPでは約15%であったのに対し,PF11-GFPでは約56%であった.このとき,PF11-GFPの蛍光は,光退色前と同じナノドメイン内に現れた.このことから,PSD-95はPSDナノドメイン内部で恒常的に局所的なパルミトイル化サイクルを受けていることが明らかとなった.我々の報告とほぼ同時期に,海外のグループからもPSDナノドメインの存在が報告された.Nairらは,四つの異なる超解像顕微鏡を用いて,AMPA受容体の挙動を観察した86).彼らは,ラット海馬初代培養神経細胞のシナプス内に,20分子程度のAMPA受容体からなる約70 nmのナノクラスターが数個含まれることを見いだした.このAMPA受容体のナノクラスターはPSD-95のナノドメインに内包されており,PSD-95の発現量によってAMPA受容体のナノクラスターの大きさが制御されていた.Broadheadらは,EGFPまたはmEOS2を融合したPSD-95をノックインしたマウスを作製し,ノックインマウス由来の海馬切片を用いて超解像顕微鏡観察を行った87).その結果,海馬切片においてもPSD-95のナノドメインの存在が示された.海馬歯状回,CA3, CA1の各層におけるPSDの大きさを比較したところ,歯状回の分子層および歯状回門,CA3の上昇層および透明層のPSDの大きさには差があり,歯状回門,透明層のPSDの方が顕著に大きかった.一方,CA1の上昇層,放射状層の近位,遠位での比較では有意な差は認められなかった.歯状回やCA3では一つのスパインに複数のナノドメインがみられることが多く,ナノドメインの数がPSDの大きさを規定しているという前述の初代培養細胞の結果と一致していた.これらの超解像顕微鏡による観察結果から,現在ではPSDナノドメインがPSDの機能をつかさどる最小構造単位であると考えられており,シナプス形成やAMPA受容体の制御におけるPSDナノドメインの重要性に注目が集まっている.さらにごく最近,シナプス前部のアクティブゾーンに,Rab3-interacting molecule(RIM)88)やMunc13-189)を含む同様のナノドメインが報告された88, 89).興味深いことに,シナプス前部,後部のナノドメインは互いに対向しており,これらの配置が精緻なシナプス伝達に重要な役割を果たしているという説(シナプスナノカラム説)が報告された88).しかし,PSDナノドメインやシナプスナノカラムがどのように形成されるかや,その機能についての研究は十分には行われておらず,今後の研究が大いに期待される.
我々は,PSDナノドメインの形成に,PSD-95のパルミトイル化サイクルが中心的な役割を果たしていると仮定している85).これまでに,我々はPSD-95のパルミトイル化酵素として,ZDHHC3/7およびZDHHC2/15の二つのZDHHCサブファミリーを同定しており13),海馬神経細胞ではZDHHC3, ZDHHC2の発現が高いことを見いだしている90).ZDHHC3は主にゴルジ体に,ZDHHC2は樹状突起内のリサイクリングエンドソーム上に局在し,ZDHHC2は神経活動阻害に応答して樹状突起内のリサイクリングエンドソームからシナプス後部膜へと輸送され,PSD-95のパルミトイル化を促進する.その結果,PSDに局在するPSD-95の増加,AMPA受容体の発現増加が起こり,神経活動低下によるシナプス伝達低下が代償される90)(シナプスの恒常的可塑性,図5B左下).一方,ラット海馬初代培養神経細胞に神経活動刺激を与えると,PSDからパルミトイル化PSD-95が減少し,AMPA受容体も減少する85)(図5B右下).これらの結果は,神経活動依存的なPSD-95のパルミトイル化と脱パルミトイル化のサイクルにより,PSDナノドメインおよびAMPA受容体のPSDにおける発現が制御されていることを示唆している.我々はPSDナノドメイン形成や脳機能におけるPSD-95のパルミトイル化サイクルの生理的意義の解明を進めている.
これまで,各種細胞株,組織を用いたパルミトーム研究により,数多くのパルミトイル化タンパク質が同定されてきた.また,ZDHHCファミリーと基質タンパク質のペアについても多くの研究がなされてきた.その甲斐あって,表2に示すように,24種類も存在するZDHHCファミリーのほとんどすべてにおいて,少なくとも一つの基質タンパク質が同定された.これらの情報は,ZDHHCファミリーの生理機能,ならびにパルミトイル化修飾の生理的意義を理解する上で必須のものであり,今後も引き続き研究が進められていくべきである.それと並行して,ZDHHCファミリーのノックアウトマウス等を用いた研究の進展が期待されるが,現時点では不十分であるといえる.すべてのZDHHCファミリーのノックアウトマウスを解析できていないことに加え,同じ基質を持つZDHHCファミリーが機能相補する可能性を考慮すると,複数のZDHHCファミリーを同時にノックアウトしたマウスの作製が不可欠である.多重ノックアウトマウスの作製は手間で時間がかかるが,幸いにも,近年のゲノム編集技術の目覚しい進歩によりそれらの敷居が低くなりつつある.今後,ノックアウトマウスの解析と,その表現型を説明するのに十分な基質タンパク質の同定を行う研究が展開されることに期待する.
また,長く分子実態が不明であった脱パルミトイル化酵素としてABHD17ファミリーが同定されたことは,この分野における近年の大きな進歩の一つといえる.ABHD17ファミリーの発現抑制やノックアウトマウスの解析を通して,これまで解析が困難であった,パルミトイル化サイクルの生理的意義について解析が可能となる.しかし,現状ではABHD17ファミリーの機能解析は十分果たされているとはいえない.我々は,ABHD17ファミリーを世界に先んじてクローニングした優位性を活かし,現在,その性状解析を進めている.また,ABHD17ファミリー以外のABHDファミリーの中に,別のパルミトイル化タンパク質を基質とする脱パルミトイル化酵素が存在するかどうか,大変興味深い.引き続き,脱パルミトイル化酵素の探索も行われていくであろう.
今後,ABHD17ファミリーを通して,パルミトイル化による機能,局在制御の研究から,パルミトイル化と脱パルミトイル化のサイクルによるダイナミックな機能変動の持つ生理的意義という新たな視点での研究につながると予想される.実際,我々はPSD-95を例として,パルミトイル化サイクルによるシナプス伝達制御機構を提唱し,その証明に取り組んでいる.このような研究が,他のパルミトイル化タンパク質によっても展開されることで,本当の意味でのパルミトイル化修飾の意義に迫れるものと思われる.
謝辞Acknowledgments
本稿で紹介した我々の研究成果は,これまで当研究室に在籍した大学院生,博士研究員,スタッフとともに行ったものである.特に,横井紀彦助教と,卒業生の関谷敦志博士は,APEGS法の開発に多大な貢献を果たした.さらに技術職員,技術支援員の皆様には多くのサポートをしていただいた.ここに感謝申し上げます.また,国内外の多くの共同研究者にも,この場を借りて感謝申し上げます.
引用文献References
1) Fukata, Y. & Fukata, M. (2010) Nat. Rev. Neurosci., 11, 161–175.
2) Levental, I., Grzybek, M., & Simons, K. (2010) Biochemistry, 49, 6305–6316.
3) Nadolski, M.J. & Linder, M.E. (2007) FEBS J., 274, 5202–5210.
4) Chamoun, Z., Mann, R.K., Nellen, D., von Kessler, D.P., Bellotto, M., Beachy, P.A., & Basler, K. (2001) Science, 293, 2080–2084.
5) Lee, J.D. & Treisman, J.E. (2001) Curr. Biol., 11, 1147–1152.
6) Buglino, J.A. & Resh, M.D. (2008) J. Biol. Chem., 283, 22076–22088.
7) Schmidt, M.F., Bracha, M., & Schlesinger, M.J. (1979) Proc. Natl. Acad. Sci. USA, 76, 1687–1691.
8) Schmidt, M.F. & Schlesinger, M.J. (1979) Cell, 17, 813–819.
9) Chamberlain, L.H. & Shipston, M.J. (2015) Physiol. Rev., 95, 341–376.
10) Bartels, D.J., Mitchell, D.A., Dong, X., & Deschenes, R.J. (1999) Mol. Cell. Biol., 19, 6775–6787.
11) Lobo, S., Greentree, W.K., Linder, M.E., & Deschenes, R.J. (2002) J. Biol. Chem., 277, 41268–41273.
12) Roth, A.F., Feng, Y., Chen, L., & Davis, N.G. (2002) J. Cell Biol., 159, 23–28.
13) Fukata, M., Fukata, Y., Adesnik, H., Nicoll, R.A., & Bredt, D.S. (2004) Neuron, 44, 987–996.
14) Keller, C.A., Yuan, X., Panzanelli, P., Martin, M.L., Alldred, M., Sassoe-Pognetto, M., & Luscher, B. (2004) J. Neurosci., 24, 5881–5891.
15) Huang, K., Yanai, A., Kang, R., Arstikaitis, P., Singaraja, R.R., Metzler, M., Mullard, A., Haigh, B., Gauthier-Campbell, C., Gutekunst, C.A., Hayden, M.R., & El-Husseini, A. (2004) Neuron, 44, 977–986.
16) Edmonds, M.J. & Morgan, A. (2014) BMC Genomics, 15, 841.
17) Bannan, B.A., Van Etten, J., Kohler, J.A., Tsoi, Y., Hansen, N.M., Sigmon, S., Fowler, E., Buff, H., Williams, T.S., Ault, J.G., Glaser, R.L., & Korey, C.A. (2008) Fly (Austin), 2, 198–214.
18) Batistic, O. (2012) Plant Physiol., 160, 1597–1612.
19) Frenal, K., Kemp, L.E., & Soldati-Favre, D. (2014) Int. J. Parasitol., 44, 121–131.
20) Rana, M.S., Kumar, P., Lee, C.J., Verardi, R., Rajashankar, K.R., & Banerjee, A. (2018) Science, 359, eaao6326.
21) Fukata, Y., Iwanaga, T., & Fukata, M. (2006) Methods, 40, 177–182.
22) Tsutsumi, R., Fukata, Y., Noritake, J., Iwanaga, T., Perez, F., & Fukata, M. (2009) Mol. Cell. Biol., 29, 435–447.
23) Oku, S., Takahashi, N., Fukata, Y., & Fukata, M. (2013) J. Biol. Chem., 288, 19816–19829.
24) Brigidi, G.S., Sun, Y., Beccano-Kelly, D., Pitman, K., Mobasser, M., Borgland, S.L., Milnerwood, A.J., & Bamji, S.X. (2014) Nat. Neurosci., 17, 522–532.
25) Chen, S., Zhu, B., Yin, C., Liu, W., Han, C., Chen, B., Liu, T., Li, X., Chen, X., Li, C., Hu, L., Zhou, J., Xu, Z.X., Gao, X., Wu, X., Goding, C.R., & Cui, R. (2017) Nature, 549, 399–403.
26) Abrami, L., Dallavilla, T., Sandoz, P.A., Demir, M., Kunz, B., Savoglidis, G., Hatzimanikatis, V., & van der Goot, F.G. (2017) eLife, 6.
27) McMichael, T.M., Zhang, L., Chemudupati, M., Hach, J.C., Kenney, A.D., Hang, H.C., & Yount, J.S. (2017) J. Biol. Chem.
28) Verardi, R., Kim, J.S., Ghirlando, R., & Banerjee, A. (2017) Structure, 25, 1337–1347.
29) Oyama, T., Miyoshi, Y., Koyama, K., Nakagawa, H., Yamori, T., Ito, T., Matsuda, H., Arakawa, H., & Nakamura, Y. (2000) Genes Chromosomes Cancer, 29, 9–15.
30) Kilpatrick, C.L., Murakami, S., Feng, M., Wu, X., Lal, R., Chen, G., Du, K., & Luscher, B. (2016) J. Biol. Chem., 291, 27371–27386.
31) Li, Y., Hu, J., Hofer, K., Wong, A.M., Cooper, J.D., Birnbaum, S.G., Hammer, R.E., & Hofmann, S.L. (2010) J. Biol. Chem., 285, 13022–13031.
32) Li, Y., Martin, B.R., Cravatt, B.F., & Hofmann, S.L. (2012) J. Biol. Chem., 287, 523–530.
33) Mukai, J., Dhilla, A., Drew, L.J., Stark, K.L., Cao, L., MacDermott, A.B., Karayiorgou, M., & Gogos, J.A. (2008) Nat. Neurosci., 11, 1302–1310.
34) Mukai, J., Liu, H., Burt, R.A., Swor, D.E., Lai, W.S., Karayiorgou, M., & Gogos, J.A. (2004) Nat. Genet., 36, 725–731.
35) Mukai, J., Tamura, M., Fenelon, K., Rosen, A.M., Spellman, T.J., Kang, R., MacDermott, A.B., Karayiorgou, M., Gordon, J.A., & Gogos, J.A. (2015) Neuron, 86, 680–695.
36) Raymond, F.L., Tarpey, P.S., Edkins, S., Tofts, C., O’Meara, S., Teague, J., Butler, A., Stevens, C., Barthorpe, S., Buck, G., Cole, J., Dicks, E., Gray, K., Halliday, K., Hills, K., Hinton, J., Jones, D., Menzies, A., Perry, J., Raine, K., Shepherd, R., Small, A., Varian, J., Widaa, S., Mallya, U., Moon, J., Luo, Y., Shaw, M., Boyle, J., Kerr, B., Turner, G., Quarrell, O., Cole, T., Easton, D.F., Wooster, R., Bobrow, M., Schwartz, C.E., Gecz, J., Stratton, M.R., & Futreal, P.A. (2007) Am. J. Hum. Genet., 80, 982–987.
37) Masurel-Paulet, A., Kalscheuer, V.M., Lebrun, N., Hu, H., Levy, F., Thauvin-Robinet, C., Darmency-Stamboul, V., El Chehadeh, S., Thevenon, J., Chancenotte, S., Ruffier-Bourdet, M., Bonnet, M., Pinoit, J.M., Huet, F., Desportes, V., Chelly, J., & Faivre, L. (2014) Am. J. Med. Genet. A., 164A, 789–795.
38) Saleem, A.N., Chen, Y.H., Baek, H.J., Hsiao, Y.W., Huang, H.W., Kao, H.J., Liu, K.M., Shen, L.F., Song, I.W., Tu, C.P., Wu, J.Y., Kikuchi, T., Justice, M.J., Yen, J.J., & Chen, Y.T. (2010) PLoS Genet., 6, e1000985.
39) Liu, K.M., Chen, Y.J., Shen, L.F., Haddad, A.N.S., Song, I.W., Chen, L.Y., Chen, Y.J., Wu, J.Y., Yen, J.J.Y., & Chen, Y.T. (2015) J. Invest. Dermatol., 135, 2603–2610.
40) Sutton, L.M., Sanders, S.S., Butland, S.L., Singaraja, R.R., Franciosi, S., Southwell, A.L., Doty, C.N., Schmidt, M.E., Mui, K.K., Kovalik, V., Young, F.B., Zhang, W., & Hayden, M.R. (2013) Hum. Mol. Genet., 22, 452–465.
41) Mansouri, M.R., Marklund, L., Gustavsson, P., Davey, E., Carlsson, B., Larsson, C., White, I., Gustavson, K.H., & Dahl, N. (2005) Eur. J. Hum. Genet., 13, 970–977.
42) Zhou, T., Li, J., Zhao, P., Liu, H., Jia, D., Jia, H., He, L., Cang, Y., Boast, S., Chen, Y.H., Thibault, H., Scherrer-Crosbie, M., Goff, S.P., & Li, B. (2015) Proc. Natl. Acad. Sci. USA, 112, 15666–15671.
43) Milnerwood, A.J., Parsons, M.P., Young, F.B., Singaraja, R.R., Franciosi, S., Volta, M., Bergeron, S., Hayden, M.R., & Raymond, L.A. (2013) Proc. Natl. Acad. Sci. USA, 110, 20296–20301.
44) Singaraja, R.R., Huang, K., Sanders, S.S., Milnerwood, A.J., Hines, R., Lerch, J.P., Franciosi, S., Drisdel, R.C., Vaid, K., Young, F.B., Doty, C., Wan, J., Bissada, N., Henkelman, R.M., Green, W.N., Davis, N.G., Raymond, L.A., & Hayden, M.R. (2011) Hum. Mol. Genet., 20, 3899–3909.
45) Sanders, S.S., Hou, J., Sutton, L.M., Garside, V.C., Mui, K.K., Singaraja, R.R., Hayden, M.R., & Hoodless, P.A. (2015) Dev. Biol., 397, 257–266.
46) Mill, P., Lee, A.W., Fukata, Y., Tsutsumi, R., Fukata, M., Keighren, M., Porter, R.M., McKie, L., Smyth, I., & Jackson, I.J. (2009) PLoS Genet., 5, e1000748.
47) Israely, I., Costa, R.M., Xie, C.W., Silva, A.J., Kosik, K.S., & Liu, X. (2004) Curr. Biol., 14, 1657–1663.
48) Zhu, J.J., Qin, Y., Zhao, M., Van Aelst, L., & Malinow, R. (2002) Cell, 110, 443–455.
49) Drisdel, R.C. & Green, W.N. (2004) Biotechniques, 36, 276–285.
50) Roth, A.F., Wan, J., Bailey, A.O., Sun, B., Kuchar, J.A., Green, W.N., Phinney, B.S., Yates, J.R. 3rd, & Davis, N.G. (2006) Cell, 125, 1003–1013.
51) Kang, R., Wan, J., Arstikaitis, P., Takahashi, H., Huang, K., Bailey, A.O., Thompson, J.X., Roth, A.F., Drisdel, R.C., Mastro, R., Green, W.N., Yates, J.R. 3rd, Davis, N.G., & El-Husseini, A. (2008) Nature, 456, 904–909.
52) Forrester, M.T., Hess, D.T., Thompson, J.W., Hultman, R., Moseley, M.A., Stamler, J.S., & Casey, P.J. (2011) J. Lipid Res., 52, 393–398.
53) Hemsley, P.A., Weimar, T., Lilley, K.S., Dupree, P., & Grierson, C.S. (2013) New Phytol., 197, 805–814.
54) Zhou, B., An, M., Freeman, M.R., & Yang, W. (2014) J. Proteomics Bioinform., 7, 256–263.
55) Yokoi, N., Fukata, Y., Sekiya, A., Murakami, T., Kobayashi, K., & Fukata, M. (2016) J. Neurosci., 36, 6431–6444.
56) Hang, H.C., Geutjes, E.J., Grotenbreg, G., Pollington, A.M., Bijlmakers, M.J., & Ploegh, H.L. (2007) J. Am. Chem. Soc., 129, 2744–2745.
57) Martin, B.R. & Cravatt, B.F. (2009) Nat. Methods, 6, 135–138.
58) Ciepla, P., Konitsiotis, A.D., Serwa, R.A., Masumoto, N., Leong, W.P., Dallman, M.J., Magee, A.I., & Tate, E.W. (2014) Chem. Sci. (Camb.), 5, 4249–4259.
59) Martin, B.R., Wang, C., Adibekian, A., Tully, S.E., & Cravatt, B.F. (2011) Nat. Methods, 9, 84–89.
60) Topinka, J.R. & Bredt, D.S. (1998) Neuron, 20, 125–134.
61) Craven, S.E., El-Husseini, A.E., & Bredt, D.S. (1999) Neuron, 22, 497–509.
62) Chen, L., Chetkovich, D.M., Petralia, R.S., Sweeney, N.T., Kawasaki, Y., Wenthold, R.J., Bredt, D.S., & Nicoll, R.A. (2000) Nature, 408, 936–943.
63) El-Husseini, A.E., Schnell, E., Chetkovich, D.M., Nicoll, R.A., & Bredt, D.S. (2000) Science, 290, 1364–1368.
64) Beique, J.C., Lin, D.T., Kang, M.G., Aizawa, H., Takamiya, K., & Huganir, R.L. (2006) Proc. Natl. Acad. Sci. USA, 103, 19535–19540.
65) Elias, G.M., Funke, L., Stein, V., Grant, S.G., Bredt, D.S., & Nicoll, R.A. (2006) Neuron, 52, 307–320.
66) Brigidi, G.S., Santyr, B., Shimell, J., Jovellar, B., & Bamji, S.X. (2015) Nat. Commun., 6, 8200.
67) Duncan, J.A. & Gilman, A.G. (1998) J. Biol. Chem., 273, 15830–15837.
68) Yeh, D.C., Duncan, J.A., Yamashita, S., & Michel, T. (1999) J. Biol. Chem., 274, 33148–33154.
69) Siegel, G., Obernosterer, G., Fiore, R., Oehmen, M., Bicker, S., Christensen, M., Khudayberdiev, S., Leuschner, P.F., Busch, C.J., Kane, C., Hubel, K., Dekker, F., Hedberg, C., Rengarajan, B., Drepper, C., Waldmann, H., Kauppinen, S., Greenberg, M.E., Draguhn, A., Rehmsmeier, M., Martinez, J., & Schratt, G.M. (2009) Nat. Cell Biol., 11, 705–716.
70) Camp, L.A. & Hofmann, S.L. (1993) J. Biol. Chem., 268, 22566–22574.
71) Verkruyse, L.A. & Hofmann, S.L. (1996) J. Biol. Chem., 271, 15831–15836.
72) Heinonen, O., Kyttala, A., Lehmus, E., Paunio, T., Peltonen, L., & Jalanko, A. (2000) Mol. Genet. Metab., 69, 123–129.
73) Kim, S.J., Zhang, Z., Sarkar, C., Tsai, P.C., Lee, Y.C., Dye, L., & Mukherjee, A.B. (2008) J. Clin. Invest., 118, 3075–3086.
74) Vesa, J., Hellsten, E., Verkruyse, L.A., Camp, L.A., Rapola, J., Santavuori, P., Hofmann, S.L., & Peltonen, L. (1995) Nature, 376, 584–587.
75) Gupta, P., Soyombo, A.A., Atashband, A., Wisniewski, K.E., Shelton, J.M., Richardson, J.A., Hammer, R.E., & Hofmann, S.L. (2001) Proc. Natl. Acad. Sci. USA, 98, 13566–13571.
76) Sarkar, C., Chandra, G., Peng, S., Zhang, Z., Liu, A., & Mukherjee, A.B. (2013) Nat. Neurosci., 16, 1608–1617.
77) Scifo, E., Szwajda, A., Soliymani, R., Pezzini, F., Bianchi, M., Dapkunas, A., Debski, J., Uusi-Rauva, K., Dadlez, M., Gingras, A.C., Tyynela, J., Simonati, A., Jalanko, A., Baumann, M.H., & Lalowski, M. (2015) J. Proteomics, 123, 42–53.
78) Lin, D.T. & Conibear, E. (2015) eLife, 4, e11306.
79) Tortosa, E., Adolfs, Y., Fukata, M., Pasterkamp, R.J., Kapitein, L.C., & Hoogenraad, C.C. (2017) Neuron, 94, 809–825 e807.
80) Wedegaertner, P.B. & Bourne, H.R. (1994) Cell, 77, 1063–1070.
81) Chisari, M., Saini, D.K., Kalyanaraman, V., & Gautam, N. (2007) J. Biol. Chem., 282, 24092–24098.
82) Rocks, O., Peyker, A., Kahms, M., Verveer, P.J., Koerner, C., Lumbierres, M., Kuhlmann, J., Waldmann, H., Wittinghofer, A., & Bastiaens, P.I. (2005) Science, 307, 1746–1752.
83) Rocks, O., Peyker, A., & Bastiaens, P.I. (2006) Curr. Opin. Cell Biol., 18, 351–357.
84) El-Husseini Ael, D., Schnell, E., Dakoji, S., Sweeney, N., Zhou, Q., Prange, O., Gauthier-Campbell, C., Aguilera-Moreno, A., Nicoll, R.A., & Bredt, D.S. (2002) Cell, 108, 849–863.
85) Fukata, Y., Dimitrov, A., Boncompain, G., Vielemeyer, O., Perez, F., & Fukata, M. (2013) J. Cell Biol., 202, 145–161.
86) Nair, D., Hosy, E., Petersen, J.D., Constals, A., Giannone, G., Choquet, D., & Sibarita, J.B. (2013) J. Neurosci., 33, 13204–13224.
87) Broadhead, M.J., Horrocks, M.H., Zhu, F., Muresan, L., Benavides-Piccione, R., DeFelipe, J., Fricker, D., Kopanitsa, M.V., Duncan, R.R., Klenerman, D., Komiyama, N.H., Lee, S.F., & Grant, S.G. (2016) Sci. Rep., 6, 24626.
88) Tang, A.H., Chen, H., Li, T.P., Metzbower, S.R., MacGillavry, H.D., & Blanpied, T.A. (2016) Nature, 536, 210–214.
89) Sakamoto, H., Ariyoshi, T., Kimpara, N., Sugao, K., Taiko, I., Takikawa, K., Asanuma, D., Namiki, S., & Hirose, K. (2018) Nat. Neurosci., 21, 41–49.
90) Noritake, J., Fukata, Y., Iwanaga, T., Hosomi, N., Tsutsumi, R., Matsuda, N., Tani, H., Iwanari, H., Mochizuki, Y., Kodama, T., Matsuura, Y., Bredt, D.S., Hamakubo, T., & Fukata, M. (2009) J. Cell Biol., 186, 147–160.
著者紹介Author Profile
 平田 哲也(ひらた てつや)
平田 哲也(ひらた てつや)自然科学研究機構生理学研究所分子細胞生理研究領域生体膜研究部門特任助教.博士(理学).
略歴1988年兵庫県に生る.2011年大阪大学理学部生物科学科卒業.16年同大学院生命機能研究科博士課程修了.14年学術振興会特別研究員(DC2),16年大阪大学微生物病研究所特任研究員(常勤)を経て,17年4月より現職.
研究テーマと抱負PSD-95のパルミトイル化サイクルの制御機構,及びその生理的意義の解明を目指して研究を進めている.タンパク質の脂質修飾により,神経機能が調節される仕組みに興味を持っている.
ウェブサイトhttps://www.nips.ac.jp/fukata/