糖鎖と聞いて生物学研究者が抱く一般的な印象といえば何だろうか.1)構造が複雑である.2)何となく大事なのはわかるが,どこがどう大事か具体的にはよくわからない.3)分析が難しいのでできれば避けて通りたい.この三つではないかと思う.そしてその印象はおおむね真実をとらえており,糖鎖を専門とする研究者でさえも解析には常に困難がつきまとう.筆者は14年にわたり,特に神経系の糖転移酵素を標的にした研究を行ってきた.本稿では,その間わかってきたこと,また現在でもこの分野で未解決の課題などについて紹介したい.
糖鎖修飾で中心的な役割を持つのは糖転移酵素である.我々ヒトの細胞は,約180種の糖転移酵素を小胞体(ER)とゴルジ体に備え,通過していくタンパク質や脂質に修飾を施す.その結果生み出される糖鎖はN型糖鎖,O型糖鎖,プロテオグリカン,糖脂質など多岐にわたり,個々の構造にも大きなバリエーションが生まれる(図1)1).筆者は特に,タンパク質上のN型糖鎖,その中でも分岐構造や糖鎖末端のユニークなエピトープに着目した研究をこれまで行ってきた.まず初めに,これらの糖鎖の発現をつかさどる糖転移酵素の活性とその発現調節機構について紹介し,次にアルツハイマー病と関わる脳の糖鎖について,そして最後にそれら糖鎖の発現や機能を追跡・阻害するための化合物についての研究内容を紹介したい.
1)糖転移酵素の活性・局在調節
前述のように,糖転移酵素は基本的にゴルジ体と小胞体に局在するが,糖タンパク質上の糖鎖に作用する酵素群は主にゴルジ体に局在している.ゴルジ体では糖転移酵素は自身の基質となるべき糖タンパク質を何らかの形で認識し,糖を転移していく.この作業が段階的に行われ,それぞれのタンパク質に固有の糖鎖構造ができ上がる(図1).ところが実際にこの糖鎖修飾がゴルジ体の中でどのように行われているか,詳しいことはいまだにあまりよくわかっていない.特に,ある糖鎖構造が特定のタンパク質にしか付加されない現象は,まだほとんどが説明できない状態にある.ショウジョウバエでは,特定の糖鎖修飾に関わる酵素群がゾーンを形成するという報告があり2),哺乳類でも同様のゴルジ体ゾーンが存在するのかもしれない.少なくとも,糖転移酵素のゴルジ体局在,さらにいえばゴルジ内ゾーンの局在制御,また酵素の活性を制御するメカニズムを明らかにしていくことが,糖鎖付加の全容を知る上で重要だと考えられる.
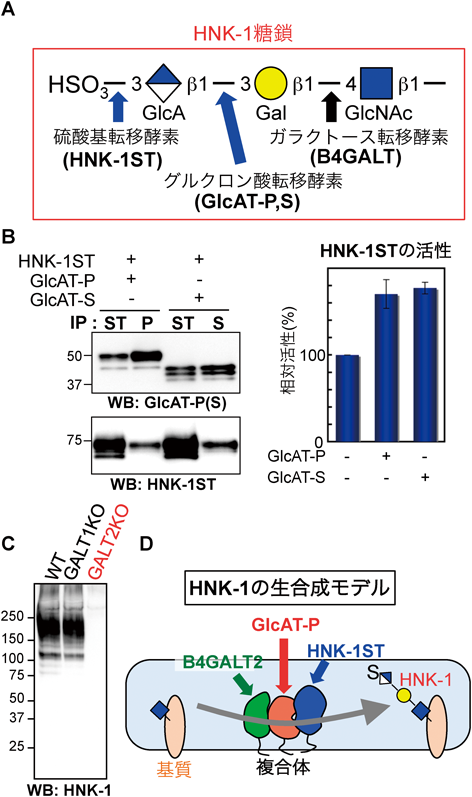
筆者が修士課程(京大・岡研究室)でHNK-1(human natural killer-1)糖鎖に関する研究をスタートさせたころ,一つの疑問があった.HNK-1糖鎖は,神経特異的な糖鎖で,N型糖鎖などの末端に存在する酸性糖鎖エピトープであり3),硫酸化されたグルクロン酸という他の糖鎖中にはない珍しい構造を有し,その生合成は糖鎖末端のガラクトースにグルクロン酸が付加され,さらにそのあと硫酸基が付加されることで行われる(図2A)4).この二つの段階は,ゴルジ体の2種の独立の酵素が行うにもかかわらず,常にグルクロン酸は硫酸化されており,硫酸がついていないものはなぜか存在しない(今では存在することがわかっているが),というのが当時の疑問であった.この疑問から,グルクロン酸転移酵素(GlcAT-P, GlcAT-Sの2種類が存在)と硫酸基転移酵素(HNK-1STという唯一の酵素)が近傍で協調的に働けるところに存在する,すなわち複合体を形成しているのではないか,という仮説を立て,それを検証すべく実験を開始した.共免疫沈降や免疫染色による解析の結果,これら二つの酵素は物理的に相互作用し,その結果,硫酸基転移活性が約2倍に上昇することがわかった(図2B).このことから,これらの酵素は実際に細胞内で複合体を形成して効率のよい糖鎖生合成を行っていると考えられた5).
さらにその後,当時金沢大学におられた浅野雅秀博士が作られたガラクトース転移酵素の欠損マウスを解析する機会をいただき,驚くべき結果が得られた.B4GALTファミリーは,β1,4結合でガラクトースを転移する酵素の一群で,それらの多くは糖鎖の共通構造の一つであるGalβ1-4GlcNAcを生合成する.1~7のアイソフォームがあり6),一部の酵素は特定の基質にのみ作用することがわかっているが,基質特異性が重複する酵素群の生体内における役割の違いについてはあまりよくわかっていない.浅野先生が作られたB4galt2欠損マウスの脳を解析すると,HNK-1の発現がきれいに消失していた(図2C)7).一方最も主要な酵素であるB4galt1の欠損では,HNK-1糖鎖の発現にはまったく変化がなかった8, 9).すなわち,生体においてはHNK-1の内部構造はB4GALT2がほぼ生合成していることを意味する.このことから,B4GALT2はHNK-1の生合成に特化した酵素である可能性も考えられた.これをふまえ,B4GALT2がGlcAT-Pと複合体を形成していると予想し,免疫沈降および免疫染色で確認したところ,B4GALT1はGlcAT-Pとほとんど相互作用しないのに対して,B4GALT2は相互作用することがわかった8).以上のことから,HNK-1生合成酵素群は,ヘテロ複合体を作って効率的な糖鎖合成を行っていることが明らかになった(図2D).
このような糖鎖合成酵素群のヘテロ複合体は他にいくつも見つかっており,酵素複合体を作ることがゴルジ体での糖鎖生合成において重要なシステムになっている可能性がある10).ただ,糖転移酵素は総じて発現量が低いために内在性の酵素の検出が難しく,生理的条件での複合体形成の解析が困難である.糖鎖合成酵素が生理的条件下のゴルジ体内でどのように複合体を形成し,それがどのように糖鎖生合成に寄与するのか,今後の解析による進展に期待したい.また糖鎖の連続的な生合成ステップから容易に想像できることであるが,糖転移酵素の細胞内における活性(非活性という意味ではない)は,酵素の微細な局在の変化によって大きく変わる.筆者が研究しているGlcAT-Pにおいても,N末端側の短い細胞質領域が13アミノ酸違うだけで局在が大きく変化し,合成されてくるHNK-1糖鎖の発現量が大きく変化する11).このような局在コントロールによる糖鎖発現制御機構も多くの糖転移酵素で見いだされており12),細胞内の糖鎖の発現パターンを知る上で重要な因子の一つになっている.糖転移酵素の局在については,多くの場合N末端側のドメインが鍵を握っており,内腔側の触媒ドメインとは独立に制御されていると考えられているが,普遍的な制御メカニズムはいまだに解明されていない.
2)糖転移酵素遺伝子のエピジェネティクス
糖転移酵素の活性や局在に加え,その発現量も糖鎖発現を規定する重要な因子である.ところが糖転移酵素の時期・組織・疾患特異的な遺伝子制御メカニズムはその多くが不明なままである.筆者は,ポスドク先の理研(谷口研究室)に赴任してから,脳に特異的に発現する糖転移酵素の遺伝子制御機構の解明に取り組んだ.GnT-IX(MGAT5B遺伝子にコードされる)は,主にO-マンノース型と呼ばれる糖鎖の分岐形成に関わっている13).またノックアウトマウスの解析から,脱髄疾患の進行に関わることが明らかになっている14).特徴的なのはその発現パターンで,脳にほぼ特異的であり,中でもニューロンで強く発現する15).しかしその細胞・組織特異的な発現をつかさどるメカニズムはほとんど不明であった.
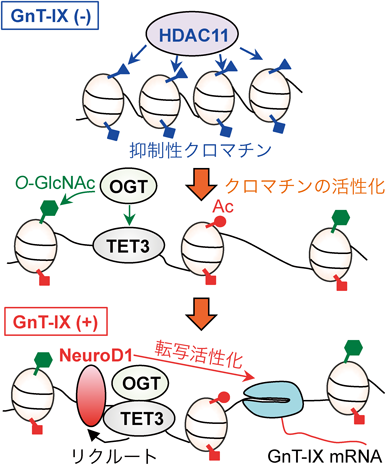
まずレポーターアッセイを用いたMgat5b遺伝子のプロモーター解析を行ったところ,Mgat5bの転写をドライブするシスエレメントが同定できた.そしてゲルシフトアッセイとChIPにより,そのエレメントに結合して転写を正に調節する二つのDNA結合因子,NeuroD1とCTCFが特定できた16).NeuroD1は脳に特異的であり,神経系の細胞株にNeuroD1を過剰発現するとGnT-IXのmRNAが大きく増加した.一方,非神経系の3T3細胞にNeuroD1を導入してもその効果は得られなかった.そこでエピジェネティックな制御を考え,3T3細胞をヒストン脱アセチル化酵素(HDAC)の阻害剤であるトリコスタチンA17)で処理したところ,Mgat5b遺伝子の転写開始部位付近のクロマチンが活性化され,発現がオンになることがわかった.さらに,Mgat5bを内在的に発現する神経系の細胞や臓器ではこの領域のクロマチンが常に活性化されており,このクロマチンの活性化によってCTCFなどのプロモーターへの結合量が制御されていることが明らかになった.これらの結果から,GnT-IXの脳特異的な発現はエピジェネティックなクロマチンの活性化状態により制御されており,組織特異的な糖鎖発現の背景にはエピジェネティクスの関与が存在することが明らかになった16).
ではMgat5b遺伝子のクロマチンはどのように活性化されているのだろうか? 次にその疑問の解決に取り組むことにした.HDAC阻害剤でその転写がオンになることから,Mgat5bが発現していない細胞では,HDACによってMgat5b遺伝子のクロマチンが抑制されている可能性が高いと考えた.そこで11種類あるHDACをそれぞれ過剰発現,およびsiRNAによるノックダウンを行ったところ,HDAC11によって選択的にMgat5bのクロマチンが抑制され,mRNAの発現が負に制御されていることがわかった15).また逆に,Mgat5bのクロマチンを活性化する因子として,OGT-TET3複合体を同定した.OGTはO-GlcNAc転移酵素であり,細胞質,ミトコンドリア,核の数多くのタンパク質に糖を転移する.ヒストンやクロマチン修飾酵素群もその基質であり,O-GlcNAc化は転写制御にも関わることが知られている18).またTET3はDNAの脱メチル化に関わる核タンパク質であり19),OGTとTETの複合体が標的遺伝子の転写を正に制御することが明らかになっている20, 21).ノックダウン実験の結果などから,OGT-TET3がMgat5b遺伝子に結合するとクロマチンが活性化され,NeuroD1などの転写活性化因子がMgat5bのプロモーターに結合しやすくなることがわかった15).さらにOGT, TET3, NeuroD1をノックダウンするとそれぞれMgat5bのmRNA量は低下した.また,HDAC11やOGT-TET3によるクロマチンの制御は遺伝子特異的であり,他の類似するGnT酵素群の遺伝子などは同様の制御を受けていないことがわかった.これらの結果から,GnT-IXの組織特異的な発現の制御に関わる因子群が明らかになってきた(図3).他にも神経特異的な糖転移酵素は多く存在しており22),それぞれの酵素が特定の転写因子,エピゲノム因子によって制御されていると考えられる.特に,クロマチンの制御やmiRNAによるエピジェネティックな糖鎖の発現制御は近年盛んに研究されており23, 24),疾患との関与も含めて今後の進展が期待される領域である.
3. N型糖鎖上のbisecting GlcNAcとアルツハイマー病(AD)
1)GnT-III(Mgat3)欠損マウスにおけるAD病態の改善
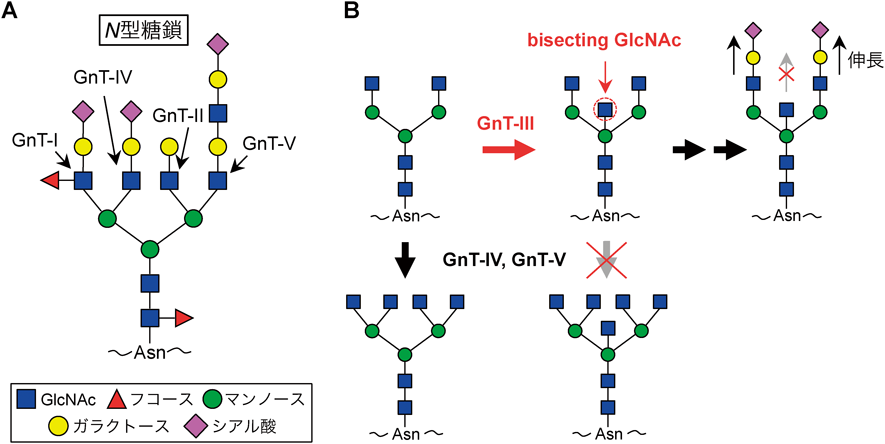
続いて疾患と関連する糖鎖の研究について紹介したい.焦点になる疾患はADであり,あるN型糖鎖の分岐構造がADの病態と関わることがわかってきた.糖タンパク質上のN型糖鎖には,三つのマンノース残基に結合したGlcNAcの数によって分岐構造が生み出され(図4A),この分岐の多寡によって糖鎖の構造と機能に多様性が生まれる.中でも,bisecting GlcNAcと呼ばれる中心の分岐構造は,神経系,特にニューロンに強く発現する構造であり,GnT-IIIと呼ばれる糖転移酵素によって生合成される(図4B)22, 25).bisecting GlcNAcは他のGlcNAc残基と機能的に大きく異なり,そこからそれ以上鎖が進展しないことや,糖鎖全体のコンホメーションを大きく変える性質があることなどが知られている26).そして,AD患者の脳でその発現が上昇することが報告され27),AD病態に何らかの関わりがあることが示唆されていた.そこで筆者らは,bisecting GlcNAcを欠損する,GnT-III(Mgat3遺伝子によりコード)欠損マウスを用いた解析を行った.
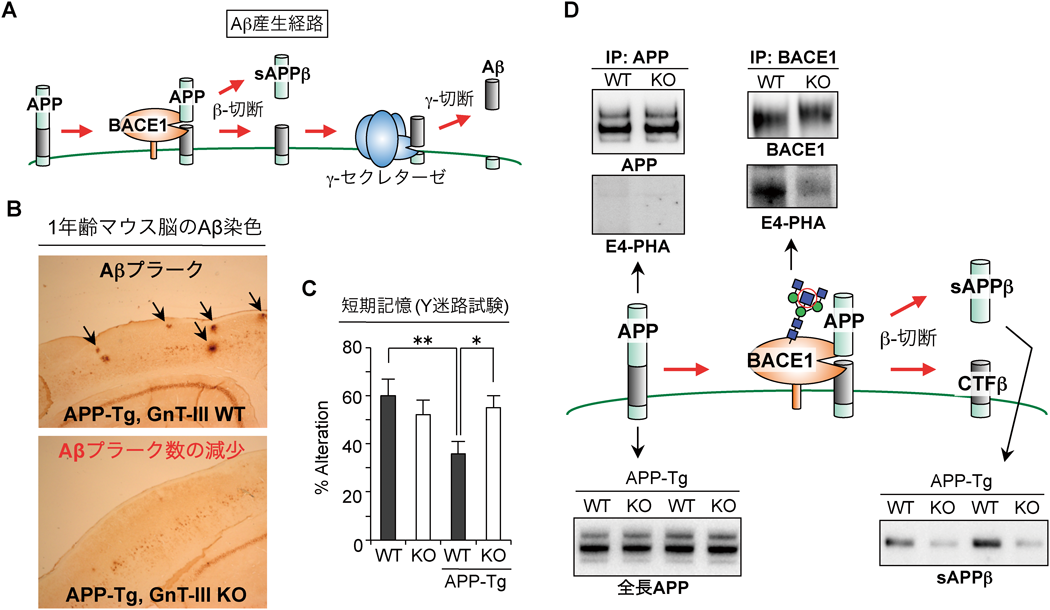
ヒトのADの二大病変は,アミロイドβ(Aβ)と呼ばれるペプチドが蓄積した老人斑(Aβプラーク)と,リン酸化タウが細胞内に蓄積した神経原線維変化である28).発症の時系列や家族性ADでみられる変異遺伝子の解析などから,現在ではAβの蓄積が病態の上流に起こるイベントであると考えられている.マウスを用いたADの研究では,Aβを蓄積するマウスがモデル動物としてよく用いられる29).AβはAPPと呼ばれる糖タンパク質が二段階の切断を受けて生じるペプチドであり,その切断はBACE1とγ-セクレターゼにより触媒される(図5A).家族性ADの変異を持ったヒトAPPのトランスジェニックマウス(以下APP-Tgマウス)はADモデルマウスとして汎用され,脳内にAβが蓄積して認知能力の低下がみられる.筆者は,APP-TgマウスとMgat3欠損マウスを交配し,このマウスにおけるADの進行を解析した.その結果,Mgat3を欠損したAPP-Tgマウスでは,Aβプラークがほとんど検出されず(図5B),Y迷路試験で測定した認知能力の低下も改善されるという驚くべき結果が得られた(図5C)30).これらの結果から,bisecting GlcNAc糖鎖がAD病態進行に深く関与すること,さらに同糖鎖が欠損すると,AD病態が改善することがわかった.
このbisecting GlcNAc糖鎖のAD進行促進メカニズムを明らかにするため,Aβ産生に関わる糖タンパク質を詳細に解析したところ,APPの一段階目の切断を担うBACE1が選択的にbisecting GlcNAc含有糖鎖を持っており,APPなどの糖タンパク質はこの糖鎖構造を持たないことがわかった(図5D上).さらにAPP切断産物のウエスタンブロットにより,Mgat3欠損マウスではBACE1によるAPPの切断が大きく低下していることがわかり(図5D下),bisecting GlcNAcはBACE1の機能を正に制御していることが示された30).次に,糖鎖による機能制御のメカニズムを探索したところ,bisecting GlcNAc修飾の有無は,BACE1の触媒活性そのものには影響を与えず,BACE1の細胞内局在を初期エンドソームから後期エンドソーム/リソソームへ変化させることがわかった.一方,APP以外のBACE1の生理的基質の切断を調べたところ,正常に切断されていた.以上のことから,bisecting GlcNAcによるBACE1の機能制御には基質特異性があることがわかった30).BACE1による基質の切断は神経機能に重要であり,BACE1欠損マウスは早生,成長遅延,ミエリン形成不全,骨格筋の異常などさまざまな表現型を呈す31, 32).一方Mgat3欠損マウスはそのような症状は示さない.以上のことから,bisecting GlcNAc合成酵素GnT-IIIを標的とした薬剤は,より副作用が少なく安全な治療薬候補になると考えられた.
2)GnT-IIIのADへの関与と阻害剤開発
ではなぜ,GnT-IIIの発現はAD患者の脳で高まるのだろうか? 筆者は酸化ストレスに着目した.GnT-IIIはBACE1と同じくストレスに応答して発現すると考えられているためである33).実際,ADでは酸化ストレスが亢進すること,培養ニューロンにAβを添加すると酸化ストレスが亢進することも知られている34, 35).Aβが蓄積したADモデルマウス(理研,西道隆臣博士より分与されたAPPノックインマウス36))の脳を染色すると,酸化ストレスマーカー,BACE1, bisecting GlcNAcのすべてが増加していた37).さらに,培養細胞にH2O2で強制的に酸化ストレスを与えたところ,BACE1上のbisecting GlcNAc量が増加し,BACE1のタンパク質発現も増加した.これをMgat3欠損細胞で行ったところ,BACE1のタンパク質量はむしろ減少し,それはクロロキンでリソソームの機能を阻害するとキャンセルされた.以上の結果から,Aβの蓄積により生じる酸化ストレスがBACE1上のbisecting GlcNAc量を増加させ,それがBACE1のリソソーム局在を抑制してタンパク質量の増加につながると考えられた37).
現在,これらの知見をふまえ,新規AD治療薬の開発を目指してGnT-IIIの阻害剤を探索している.糖転移酵素の特異的な阻害剤は,スクリーニング系の不足などからこれまでほとんど開発されていない.筆者らは,糖転移反応の副産物であるUDPを定量する系を用いて新たなスクリーニング系を構築し,現在化合物ライブラリを用いてスクリーニングを行っている.
1)糖鎖の高感度クリックプローブの開発
少し話は変わるが,これまで糖鎖の研究を行ってきて常々感じていることは,ツールの不足である.糖転移酵素については,各種キナーゼに対するような特異的な阻害剤がそろっているわけでもなし,糖鎖を可視化するGFPのような改変可能プローブもない.より簡便に糖鎖の機能を改変したり追跡できたりするツールが開発されれば,糖鎖生物学は飛躍的に進歩するのではないかと思う.そんな中,研究グループ内の有機化学者との共同研究で,思わぬ形で一つのプローブの開発につながる研究を行うことができた.以下にその研究例を紹介したい.
クリックケミストリーを組み合わせた糖鎖のケミカルバイオロジー解析は,Bertozziらのグループの成果を中心にここ10年で大きく進展した38, 39).この手法では,アジド基やアルキンを有する単糖を細胞に添加し,細胞中の糖鎖に取り込ませたのち,糖鎖を化学的に検出する(図6A).原理は明快で非常に汎用性が高いようにも思えるが,特異性や産物の詳細な同定の不足など,さまざまな問題点がまだある.
現在この手法が適用可能な糖は,シアル酸,GalNAc(GlcNAc),フコースである.特にフコースは糖鎖の末端部分に位置し,特徴的なエピトープを形成しやすく,さまざまな疾患と深く関わることが知られている40, 41).糖鎖の根元のコアフコース,非還元末端側のルイス型フコース,Notchなどの一部のタンパク質に直接結合したO-フコースなどが哺乳類では知られている.フコース含有糖鎖を検出するための,6-アジドフコースと6-アルキニルフコース(Fucose alkyneの名でThermoFisherから売られている)が開発されていた42–44).しかし6-アジドフコースは細胞毒性があること,6-アルキニルフコースは感度が低いことも知られていた.
筆者が所属していた谷口グループでは,糖の合成を専門とする化学者がおり,さまざまなフコース誘導体を作り出していた(図6B).筆者は,これらの中からよりよい糖鎖プローブを探索することにした.フコース含有糖鎖構造の中で最もメジャーなものはコアフコースであり,この構造中のフコースはFUT8により付加されるが45),これまでフコースプローブとして用いられていた6-アルキニルフコースは,天然のフコースに比べてはるかにFUT8の基質として使われにくいことがわかり,これが検出感度の低い原因ではないかと考えられた(図6C).一方,新たに開発されたアルキン型フコースの中で,7-アルキニルフコースは,FUT8の基質として適していることがわかった(図6C)46).さらに7-アルキニルフコースは他のフコース転移酵素にも利用されやすいという結果が得られたことから,7-アルキニルフコースは糖鎖中に取り込まれやすく,高感度で糖鎖を検出するプローブになると考えられた.実際7-アルキニルフコースを細胞に添加したところ,6-アルキニルフコースに比べて感度よく糖鎖を検出できることがわかった(図6D)46).また,6-アジドフコースのような毒性もみられなかった.
添加した7-アルキニルフコースがどのように糖鎖中に取り込まれるかを解析するため,藤田保健衛生大の中嶋和紀博士,広島大の中の三弥子博士らと共同で糖ヌクレオチドと糖鎖を分析した.その結果,培地中に添加した7-アルキニルフコースは細胞内でGDP体へ変換され,糖鎖中に主にコアフコースの形で取り込まれることがわかった.以上の結果から,新たに開発したフコースアナログである7-アルキニルフコースは,感度のよい新しいフコシル化糖鎖プローブであることが明らかになった.
2)糖鎖合成を阻害する糖アナログの開発
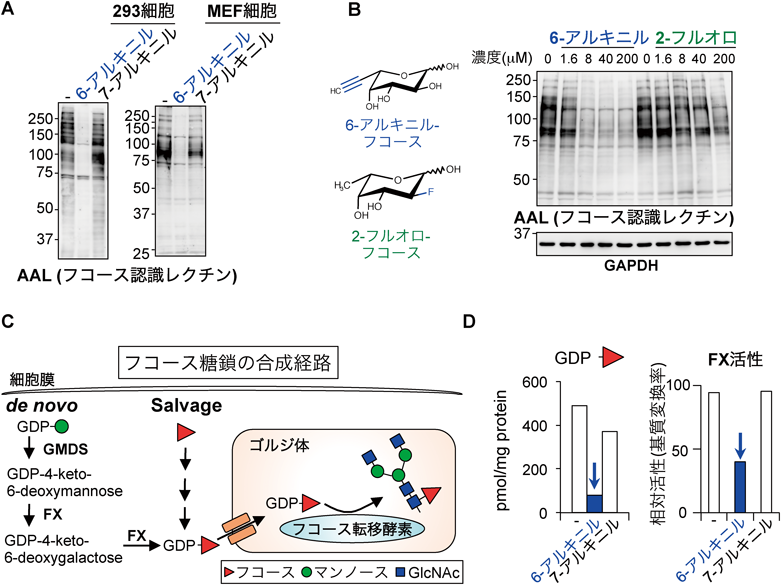
上述のフコース誘導体の研究の過程で,一つ面白い発見があった.対照として使っていた6-アルキニルフコースであるが,これを添加した細胞ではいつもフコース認識レクチンとの反応性が悪くなるということである(図7A).これはすなわち,6-アルキニルフコースは内在性のフコース含有糖鎖の発現を阻害しているということを意味する.一方で,7-アルキニルフコースではそのような効果はみられなかった.質量分析でより詳細なグライコミクス解析を行ったところ,フコースを一つ持った糖鎖や二つ持った糖鎖の発現が6-アルキニルフコース処理細胞ではほとんどなくなり,代わりに対応するフコース非含有糖鎖が大きく増加していた47).これらの結果から,やはり6-アルキニルフコースは内在性のフコシル化を阻害していると考えられた.さらに,その阻害効果は既存の阻害剤である2-フルオロフコースよりも有意に高いことがわかった(図7B).
次の疑問は,阻害のメカニズムである.それを知るには,細胞内のフコシル化の経路について考える必要がある.動物細胞は,グルコースを出発原料として,GDP-マンノースを経てGDP-フコースを作るde novo経路と,遊離のフコースを再利用するsalvage経路があるが(図7C),ほぼde novo経路に依存している40).GDP-マンノースはGMDS, FXと呼ばれる二つの酵素によりGDP-フコースへと変換され,その後GDP-フコーストランスポーターによってゴルジ体内腔側へと輸送され,フコース転移酵素によって糖鎖中にフコースが取り込まれる.まず,6-アルキニルフコースがフコース転移酵素を阻害している可能性を検証したが,in vitroにおけるフコース転移酵素アッセイでは,阻害効果は確認できなかった.そこでGDP-フコースに目をつけた.6-アルキニルフコースで処理した細胞内のGDP-フコースを定量したところ,ほぼ枯渇していることがわかった(図7D).このことから,6-アルキニルフコースは,GDP-フコース合成酵素を阻害している可能性が強く示唆された.そこで,GMDSとFXの酵素活性測定系に6-アルキニルフコースを添加したところ,FXが選択的にかつ強力に阻害されることがわかった(図7D).以上のことから,6-アルキニルフコースはFXをブロックすることでGDP-フコース量を減少させ,内在性のフコシル化を起こらなくさせることが明らかになった47).さらに,この作用によって細胞のフコシル化を抑えると,肝がん細胞の浸潤能を低下させることもわかった.このことは,フコシル化糖鎖ががんの悪性化に関わっており,糖鎖の発現を変化させる化合物が疾患の治療候補物質になりうることを示している.
以上の結果から,糖アナログの糖鎖研究ツールとしての有用性がみえてきた.今後もさまざまなアナログが開発されれば,より選択性の高いプローブや強力な阻害剤の開発につながり,それらを用いた基礎研究・臨床応用の発展が期待できるだろう.
学部4年で糖鎖生物学の世界に踏み込んでから,そろそろ14年がたとうとしている.その間,糖鎖の持つ不思議な仕組みを何とか明らかにしようと思い,いろいろなことにチャレンジしてきた.しかし,何か少し答えらしきものがみえて一つの論文を出すたび,それ以上の数の新しい疑問が生まれ続け,課題は増える一方である.
糖鎖に関しては二つ大きな疑問が残っていると感じる.一つは発現の制御であり,やはりゴルジ体の中で起こっている糖鎖付加反応の現場を詳細に見たいという個人的な願望がある.おそらく,ランダム(確率論的)に起こることと,規則に従って起こることが混在しており,それを明らかにしない限り,各タンパク質上の糖鎖のバリエーションの違いが生まれるメカニズムは説明できないだろう.もう一つは物理的な糖鎖の機能である.糖鎖の構造バリエーションの大きさに対して,生体内で同定されている糖鎖認識タンパク質は少なすぎ,何をしているのか不明な糖鎖構造が多数存在している.もちろんシアル酸による電荷,分岐によるかさ高さの増加,水溶性の付加などさまざまな物理化学的性質の変化もあるだろう.各糖鎖が一体何をしているのか,それはきっと各糖鎖の正確な三次元構造を明らかにし,計算科学の力を借りなければわからないだろう.そうした課題に対して,自分にできる生化学でこれからどこまで迫れるかわからないが,新しい糖鎖生物学の境地にいつか到達できることを目指して進んでいきたいと願っている.
謝辞Acknowledgments
本研究は多くの方々のご協力により遂行したものです.HNK-1糖鎖に関する研究は京都大学の岡昌吾先生,エピジェネティクス,ADおよび糖アナログに関する研究は理化学研究所の谷口直之先生の研究室で行いました.両先生には実験のてほどきだけでなく,論文指導や研究者としてのあり方など,今の自分の核となる部分のご指導をいただきました.深く感謝申し上げます.またこれまで多くの共同研究者の先生方に多大なご協力をいただきました.紙面の都合上お一人ずつのお名前は割愛させていただきますことをお赦しください.また京都大学大学院薬学研究科生体分子認識学分野の皆様,理化学研究所疾患糖鎖研チームの皆様には日々多くのご助言とお力添えをいただきました.この場を借りて感謝申し上げます.
引用文献References
1) Varki, A. (2017) Glycobiology, 27, 3–49.
2) Yano, H., Yamamoto-Hino, M., Abe, M., Kuwahara, R., Haraguchi, S., Kusaka, I., Awano, W., Kinoshita-Toyoda, A., Toyoda, H., & Goto, S. (2005) Proc. Natl. Acad. Sci. USA, 102, 13467–13472.
3) Kizuka, Y. & Oka, S. (2012) Cell. Mol. Life Sci., 69, 4135–4147.
4) Morise, J., Takematsu, H., & Oka, S. (2017) Biochim. Biophys. Acta, 1861, 2455–2461.
5) Kizuka, Y., Matsui, T., Takematsu, H., Kozutsumi, Y., Kawasaki, T., & Oka, S. (2006) J. Biol. Chem., 281, 13644–13651.
6) Hennet, T. (2002) Cell. Mol. Life Sci., 59, 1081–1095.
7) Yoshihara, T., Sugihara, K., Kizuka, Y., Oka, S., & Asano, M. (2009) J. Biol. Chem., 284, 12550–12561.
8) Kouno, T., Kizuka, Y., Nakagawa, N., Yoshihara, T., Asano, M., & Oka, S. (2011) J. Biol. Chem., 286, 31337–31346.
9) Kido, M., Asano, M., Iwakura, Y., Ichinose, M., Miki, K., & Furukawa, K. (1998) Biochem. Biophys. Res. Commun., 245, 860–864.
10) Kellokumpu, S., Hassinen, A., & Glumoff, T. (2016) Cell. Mol. Life Sci., 73, 305–325.
11) Kizuka, Y., Tonoyama, Y., & Oka, S. (2009) J. Biol. Chem., 284, 9247–9256.
12) Tu, L. & Banfield, D.K. (2010) Cell. Mol. Life Sci., 67, 29–41.
13) Inamori, K., Endo, T., Gu, J., Matsuo, I., Ito, Y., Fujii, S., Iwasaki, H., Narimatsu, H., Miyoshi, E., Honke, K., & Taniguchi, N. (2004) J. Biol. Chem., 279, 2337–2340.
14) Kanekiyo, K., Inamori, K., Kitazume, S., Sato, K., Maeda, J., Higuchi, M., Kizuka, Y., Korekane, H., Matsuo, I., Honke, K., & Taniguchi, N. (2013) J. Neurosci., 33, 10037–10047.
15) Kizuka, Y., Kitazume, S., Okahara, K., Villagra, A., Sotomayor, E.M., & Taniguchi, N. (2014) J. Biol. Chem., 289, 11253–11261.
16) Kizuka, Y., Kitazume, S., Yoshida, M., & Taniguchi, N. (2011) J. Biol. Chem., 286, 31875–31884.
17) Yoshida, M., Kijima, M., Akita, M., & Beppu, T. (1990) J. Biol. Chem., 265, 17174–17179.
18) Hardiville, S. & Hart, G.W. (2014) Cell Metab., 20, 208–213.
19) Wu, X. & Zhang, Y. (2017) Nat. Rev. Genet., 18, 517–534.
20) Chen, Q., Chen, Y., Bian, C., Fujiki, R., & Yu, X. (2013) Nature, 493, 561–564.
21) Vella, P., Scelfo, A., Jammula, S., Chiacchiera, F., Williams, K., Cuomo, A., Roberto, A., Christensen, J., Bonaldi, T., Helin, K., & Pasini, D. (2013) Mol. Cell, 49, 645–656.
22) Kizuka, Y., Nakano, M., Miura, Y., & Taniguchi, N. (2016) Proteomics, 16, 2854–2863.
23) Lauc, G., Vojta, A., & Zoldos, V. (2014) Biochim. Biophys. Acta, 1840, 65–70.
24) Kurcon, T., Liu, Z., Paradkar, A.V., Vaiana, C.A., Koppolu, S., Agrawal, P., & Mahal, L.K. (2015) Proc. Natl. Acad. Sci. USA, 112, 7327–7332.
25) Nishikawa, A., Ihara, Y., Hatakeyama, M., Kangawa, K., & Taniguchi, N. (1992) J. Biol. Chem., 267, 18199–18204.
26) Kizuka, Y. & Taniguchi, N. (2016) Biomolecules, 6, E25.
27) Akasaka-Manya, K., Manya, H., Sakurai, Y., Wojczyk, B.S., Kozutsumi, Y., Saito, Y., Taniguchi, N., Murayama, S., Spitalnik, S.L., & Endo, T. (2010) Glycobiology, 20, 99–106.
28) Scheltens, P., Blennow, K., Breteler, M.M., de Strooper, B., Frisoni, G.B., Salloway, S., & Van der Flier, W.M. (2016) Lancet, 388, 505–517.
29) LaFerla, F.M. & Green, K.N. (2012) Cold Spring Harb. Perspect. Med., 2, a006320.
30) Kizuka, Y., Kitazume, S., Fujinawa, R., Saito, T., Iwata, N., Saido, T.C., Nakano, M., Yamaguchi, Y., Hashimoto, Y., Staufenbiel, M., Hatsuta, H., Murayama, S., Manya, H., Endo, T., & Taniguchi, N. (2015) EMBO Mol. Med., 7, 175–189.
31) Savonenko, A.V., Melnikova, T., Laird, F.M., Stewart, K.A., Price, D.L., & Wong, P.C. (2008) Proc. Natl. Acad. Sci. USA, 105, 5585–5590.
32) Willem, M., Garratt, A.N., Novak, B., Citron, M., Kaufmann, S., Rittger, A., DeStrooper, B., Saftig, P., Birchmeier, C., & Haass, C. (2006) Science, 314, 664–666.
33) Taniguchi, N., Kizuka, Y., Takamatsu, S., Miyoshi, E., Gao, C., Suzuki, K., Kitazume, S., & Ohtsubo, K. (2016) Arch. Biochem. Biophys., 595, 72–80.
34) Kao, S.C., Krichevsky, A.M., Kosik, K.S., & Tsai, L.H. (2004) J. Biol. Chem., 279, 1942–1949.
35) Mohmmad Abdul, H., Sultana, R., Keller, J.N., St Clair, D.K., Markesbery, W.R., & Butterfield, D.A. (2006) J. Neurochem., 96, 1322–1335.
36) Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., Iwata, N., & Saido, T.C. (2014) Nat. Neurosci., 17, 661–663.
37) Kizuka, Y., Nakano, M., Kitazume, S., Saito, T., Saido, T.C., & Taniguchi, N. (2016) Biochem. J., 473, 21–30.
38) Laughlin, S.T. & Bertozzi, C.R. (2009) Proc. Natl. Acad. Sci. USA, 106, 12–17.
39) Rouhanifard, S.H., Nordstrom, L.U., Zheng, T., & Wu, P. (2013) Chem. Soc. Rev., 42, 4284–4296.
40) Becker, D.J. & Lowe, J.B. (2003) Glycobiology, 13, 41R–53R.
41) Schneider, M., Al-Shareffi, E., & Haltiwanger, R.S. (2017) Glycobiology, 27, 601–618.
42) Hsu, T.L., Hanson, S.R., Kishikawa, K., Wang, S.K., Sawa, M., & Wong, C.H. (2007) Proc. Natl. Acad. Sci. USA, 104, 2614–2619.
43) Sawa, M., Hsu, T.L., Itoh, T., Sugiyama, M., Hanson, S.R., Vogt, P.K., & Wong, C.H. (2006) Proc. Natl. Acad. Sci. USA, 103, 12371–12376.
44) Rabuka, D., Hubbard, S.C., Laughlin, S.T., Argade, S.P., & Bertozzi, C.R. (2006) J. Am. Chem. Soc., 128, 12078–12079.
45) Uozumi, N., Yanagidani, S., Miyoshi, E., Ihara, Y., Sakuma, T., Gao, C.X., Teshima, T., Fujii, S., Shiba, T., & Taniguchi, N. (1996) J. Biol. Chem., 271, 27810–27817.
46) Kizuka, Y., Funayama, S., Shogomori, H., Nakano, M., Nakajima, K., Oka, R., Kitazume, S., Yamaguchi, Y., Sano, M., Korekane, H., Hsu, T.L., Lee, H.Y., Wong, C.H., & Taniguchi, N. (2016) Cell. Chem. Biol., 23, 782–792.
47) Kizuka, Y., Nakano, M., Yamaguchi, Y., Nakajima, K., Oka, R., Sato, K., Ren, C.T., Hsu, T.L., Wong, C.H., & Taniguchi, N. (2017) Cell. Chem. Biol.
48) 木塚康彦(2015)生化学,87, 381–384.
49) 木塚康彦,北爪しのぶ,谷口直之(2017)生化学,89, 626–633.
著者紹介Author Profile
 木塚 康彦(きづか やすひこ)
木塚 康彦(きづか やすひこ)岐阜大学生命の鎖統合研究センター准教授.博士(薬学).
略歴2004年京都大学薬学部卒.09年同大学院薬学研究科博士課程修了.同年4月より理化学研究所疾患糖鎖研究チーム,特別研究員.同所,基礎科学特別研究員,研究員を経て17年10月より現職.
研究テーマと抱負神経系に発現するユニークな糖鎖を対象に,複雑な糖鎖の発現制御メカニズムと,いまだに不明な点が多い糖鎖の物理的な機能を解明したいと思っています.
趣味フットサル,読書,料理.