免疫系は,病原体感染から我々の身体を守るために高度に構築されたシステムであり,自然免疫受容体が病原体成分を認識することで一連の応答が開始される.一方,これらの自然免疫受容体は病原体のみならず損傷自己に由来する成分を認識して免疫応答を惹起することで,組織恒常性維持に寄与していると考えられている1, 2).これらの認識に関わるパターン認識受容体(pattern recognition receptors:PRRs)として,Toll-like receptors(TLRs),Nod-like receptors(NLRs),RIG-I-like receptors(RLRs)が知られているが3, 4),近年これらに加えてC型レクチン受容体(C-type lectin receptors:CLRs)に関する研究が特に進展し,さまざまな免疫応答において重要な役割を担うファミリーであることが明らかとなってきた.Macrophage inducible C-type lectin(Mincle/Clec4e)はこのCLRファミリーに属し,さまざまな刺激でNF-IL6(CEBP/β)依存的に発現が誘導される分子として,1999年に松本らによって同定された5).Mincleは,糖脂質を認識することが示された最初の活性化CLRである.
2. Mincleは組織損傷を感知し免疫を活性化する受容体である
CLRsの中には,T細胞受容体やB細胞受容体のシグナル伝達に用いられることが知られているITAM(immunoreceptor tyrosine-based activation motif)(YxxL(x)6~8YxxL)を介してシグナルを伝達するものが存在する.ミエロイド細胞内においてこのシグナル伝達は,CARD9, Malt1, Bcl10の複合体形成とそれに続く,NF-κBの活性化を引き起こす.この際,CLRsが自身の細胞内領域に一つのYxxL配列からなるhemITAMモチーフを有する場合と,CLRsにITAMを含有するアダプター分子が会合する場合がある.Mincleは,細胞内領域にシグナル伝達モチーフを持たないが,細胞膜領域に正電荷アミノ酸であるアルギニンを有しており,アダプター分子と結合しシグナルを伝達することが想定された.そこでITAMを有するアダプター分子について,免疫沈降法によってMincleとの結合を調べたところ,FcRγと特異的に会合することが判明した6).FcRγはFcγR, FcεRIのみならずさまざまな受容体と結合し,ITAMのリン酸化を介して活性化シグナルを伝達することが知られている.実際,マクロファージを抗Mincle抗体で刺激すると,炎症性サイトカイン,ケモカインの産生が認められ,その産生はFcRγを欠損することで消失したことから,MincleはFcRγを介したシグナル伝達によって炎症応答を促進する活性化受容体であることが示唆された.
では,Mincleは何を認識しているのであろうか.そこで,NFAT-GFPレポーター細胞7)にMincleとFcRγを発現させ,リガンド認識を蛍光で検出する細胞を樹立した.この細胞をたまたま培地を変えずに数日間培養すると,死細胞の増加に伴い,GFPの発現が誘導されることが判明した.このレポーター活性は,死細胞を刺激に用いた際にも濃度依存的に認められたことから,Mincleが死細胞を認識すると示唆された.死細胞はマクロファージからの炎症性サイトカイン産生を誘導したが,この効果は抗Mincle抗体によって阻害された.さらにin vivoで胸腺細胞死を誘導した際に観察される胸腺への好中球浸潤も,抗Mincle抗体によって抑制されることがわかった.以上の一連の実験結果から,Mincleは損傷自己を感知し,炎症反応を惹起する受容体であることが示唆された6).
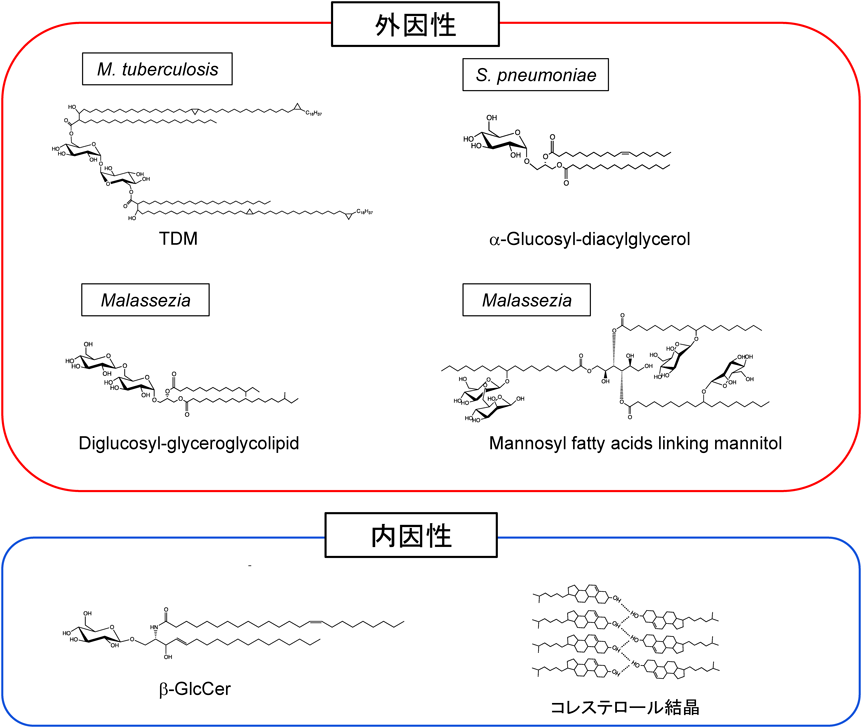
PRRsは一般に病原体を認識する.上述のMincleレポーター細胞を様々な細菌で刺激したところ,結核菌がMincleレポーター細胞を活性化することが判明した.結核菌成分を水溶性,脂溶性に分画すると,脂溶性成分でレポーター活性が認められ,この脂溶性成分を高性能薄層クロマトグラフィー(HPTLC)によってさらに分画した結果,一つのフラクションにおいて強い活性が認められ,最終的に活性を担う分子は,トレハロースジミコール酸(trehalose 6,6′-dimycolate:TDM)と呼ばれる糖脂質であることが判明した.結核菌は細胞壁に特徴的な糖脂質を有する病原体である.そのうちTDMは,古くからその免疫賦活化作用が注目されていた細胞壁糖脂質であり,完全フロイントアジュバント(complete Freund’s adjuvant : CFA)の主要な構成成分の一つとしても知られている.しかし,この分子を認識する受容体はTDMの発見後50年以上にわたり未同定であった.TDMによるマクロファージからの炎症性サイトカイン産生は,Mincleを欠損させると抑制され,MincleがTDMによる自然免疫活性化を担うことが明らかとなった8).さらに我々は,TDMを基に合成された人工アジュバントであるtrehalose dibehenate(TDB)もMincleが認識することを見いだした.
Schoenenらは,野生型とMincle欠損マウスにこのTDBをアジュバントとして免疫を行い,アジュバント活性がMincleに完全に依存することを示している9).我々のグループにおいてもTDMによる獲得免疫の活性化がMincleに依存することを,in vitroではOT-IIトランスジェニックマウス由来T細胞と骨髄由来樹状細胞(bone marrow-derived dendritic cell:BMDC)の共培養の系によって,in vivoでは遅延型過敏反応(DTH)を観察することで証明している10).これらの結果から,Mincleが糖脂質アジュバントの生物活性を担う機能的な受容体であることが明らかとなった.
一般にC型レクチン受容体は細胞外領域の糖認識ドメイン(carbohydrate recognition domain:CRD)を用いてCa2+依存的に糖鎖を認識すると考えられている.CRD内のEPN(Glu-Pro-Asn)配列はMincleを含め,グルコースやマンノースを認識するCLRsに多く見られる11).MincleとTDMの結合はEPN領域の変異によって阻害されるため,この配列を介してTDMと結合していると推測されるが,なぜTDMがMincleのみに認識され,他のEPN配列を持つCLRsには認識されないのか,詳細は不明であった.
トレハロースは,グルコースがα-1,1結合した二糖である.FeinbergらはMincleのCRDが単糖グルコースよりもトレハロースと強く結合することを明らかにした12).結晶構造解析により,Mincleは他のCLRsでもみられるCRD内のCa2+結合領域に隣接した糖結合ポケットに加え,近傍にもう一つの糖結合ポケットを有することがわかった12, 13).後者の結合領域のアミノ酸を置換すると,MincleのTDMとの結合能は減少したことから,二つ目の糖結合ポケットでの結合がMincleによるTDM認識を担うことが示された.この糖結合ポケットはMincle特異的に認められ,Mincleとリガンドの結合を増強することから,Mincleは二糖分子との結合に適した構造を有していることが推測された.
さらに構造解析により,Mincleが特徴的な疎水性の溝を有することが明らかになった12).TDMは糖構造に2本のミコール酸側鎖が付加した分子である.ミコール酸は長鎖脂肪酸であることから,Mincleの疎水性領域と相互作用する可能性が考えられた.実際,Mincleの疎水性の溝を形成するアミノ酸を置換すると,結合能の低下が認められ,この領域がMincleとTDMの結合に寄与することが示唆された.同時期に,古川,森らによってもこのことは証明されている14).加えて,Feinbergらは,トレハロースの片方のグルコース構造にアシル鎖を付加し,伸長していくとMincleとの結合が増強することを確認した12, 15).以上よりMincleは,二つの糖結合ポケットと疎水性の溝を有する,両親媒性の糖脂質認識に適した受容体であることが明らかとなった.
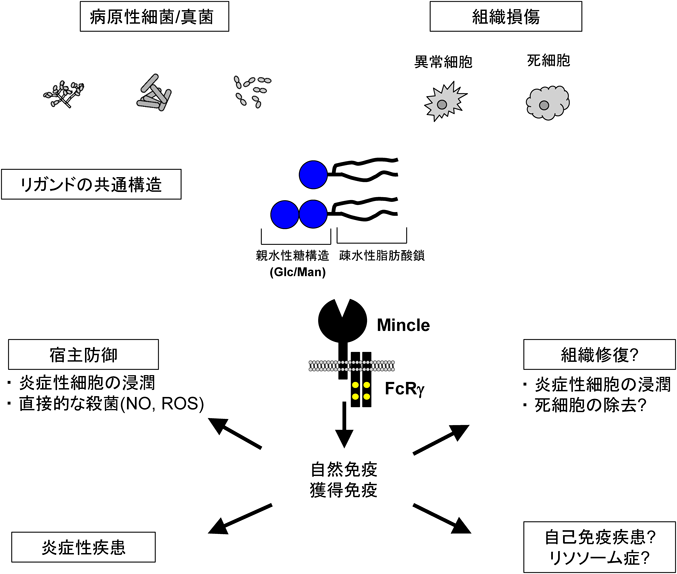
Mincleはマラセチアや肺炎球菌由来の糖脂質を認識することも報告されている16, 17).これら病原体の糖脂質が誘導する免疫応答は,TDMの結果と同様にMincleに依存した.これらの報告から明らかとなったリガンドはどれも,親水性の糖に長い疎水性脂肪酸鎖がついたものであり,Mincleの構造解析の結果と合致していた.実際,MincleとTDMの結合様式が結晶構造解析によって解明されたのち,さまざまなMincleリガンドの構造類似体が合成された18–20).これら多くのリガンドとの相互作用の研究から,Mincleの細胞外領域に含まれる,二つの糖結合ポケットと二つの疎水性領域の四つのリガンド認識モチーフのうち,少なくとも三つを満たす糖脂質はMincleのリガンドとなりうることも示唆されており20),今後,Mincleをターゲットとした新たなアジュバントをデザインするための構造的根拠が整ってきた.
5. Mincleは内因性糖脂質β-GlcCerを認識する
前述のように,Mincleを発現させたレポーター細胞は死細胞増加に伴い活性化する.Mincleが糖脂質を特異的に認識することから,死細胞上清中に含まれる何らかの内因性糖脂質成分がこのリガンド活性を担う可能性が想定された.そこで,この上清から抽出した脂質成分を,高速液体クロマトグラフィー(HPLC)により分画し,レポーター細胞を用いてリガンド活性を追跡したところ,一つのフラクションにおいて強い活性が死細胞上清でのみ検出された(図1A, B).このフラクションにはいくつかの成分が混在していたことから,さらにHPTLCにより精製を試み,強いMincleリガンド活性を有するサブフラクションを同定した(図1C, D).九州大学生体防御医学研究所メタボロミクス分野の馬場,和泉らとの共同研究により,超臨界流体クロマトグラフィー/質量分析(SFC-MS)を用いてこの成分の分子量を計測し(図1E),代謝プロファイルデータベース21)と照合した結果,β-GlcCer,またはβ-ガラクトシルセラミド(β-GalCer)が活性本体であることが強く示唆された.β-GlcCerがすべての細胞に普遍的に存在するのに対し,β-GalCerは主にミエリン鞘を構成する糖脂質であることから,リガンド活性を担う成分はβ-GlcCerであることが推測された.実際,細胞損傷に伴って得られるMincleレポーター活性は,β-GlcCer合成酵素(UDP-glucose ceramide glucosyltransferase:UGCG)の阻害剤(D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol:D-PDMP)を加えることで失われ,この系においても死細胞由来のβ-GlcCerがMincleリガンド活性を担っていたことが強く示唆された.
β-GlcCerと推測されるMincleリガンド活性成分は,生細胞からも得られた.この成分は,死細胞上清から得られるリガンド成分と同一であることがMS解析によって判明した.このことは,このリガンド成分が,細胞死に伴い発現が誘導されるものではなく,細胞内に恒常的に存在するものであることを示した.我々は,生細胞から得られる大量のリガンド成分を用いて,九州大学薬学研究院臨床薬学部門の宮本との共同研究により,核磁気共鳴法(NMR)を行った.この結果,分子量だけでは決定できなかった糖構造がβ-グルコースであることが確認され,Mincle内因性リガンドがβ-GlcCerと同定された.実際,合成β-GlcCerによってMincleレポーター細胞を刺激したところ,レポーター遺伝子の発現が認められ,β-GlcCerがMincleの内因性リガンドであることが証明された22).
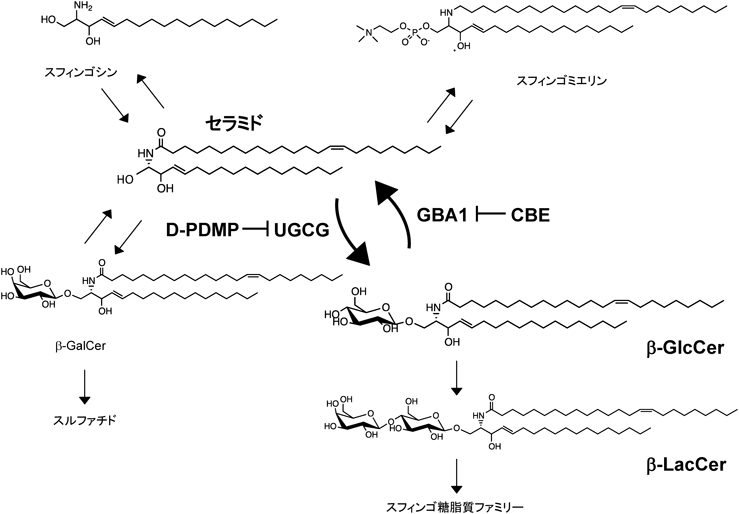
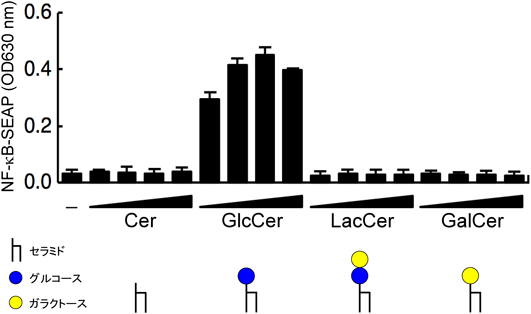
β-GlcCerは,セラミドから始まるスフィンゴ糖脂質代謝経路のうち,最初に合成される糖脂質であり(図2)23),その後のスフィンゴ糖脂質ファミリー生合成に必須な中間代謝産物である.我々は,Mincleによる内因性糖脂質認識がβ-GlcCer特異的であるか検討を行った.β-GlcCerの上流,下流にあるセラミド,ラクトシルセラミド(LacCer),さらにセラミドから合成される別の糖脂質であるβ-GalCerはMincleレポーター細胞をまったく活性化せず,これら類似する糖脂質の中で,β-GlcCerが唯一Mincleに認識されることが示唆された(図3).
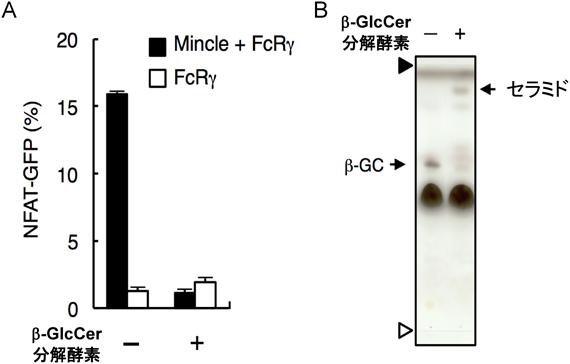
β-GlcCerは,これまでにNKT細胞によっても認識されることが報告されていた24).しかし,その報告の3年後には,そのリガンド活性は少量混入していたα-GlcCerがもたらしていたことが報告された25–27).今回のMincleリガンド活性が,混入物によってもたらされていた可能性を除去するため,β-GlcCer分解酵素処理を行ったところ,β-GlcCerの分解に伴いリガンド活性が失われることが観察され,β-GlcCerが真のMincleリガンドであることが証明された(図4A, B).セラミドを認識する抑制性の受容体としてLMIR3も見つかっている28).一方β-GalCerもまた,type-II NKT細胞を介して抑制性に働くことが示唆されている29).β-GlcCerはセラミド代謝経路における初めての免疫賦活化物質である.
今回我々は,Mincleリガンド活性を有するフラクションから脂肪酸鎖が異なる6種類のβ-GlcCerを同定した[C16:0, C18:0, C20:0, C22:0, C24:0, C24:1(15Z)](図1E).このうち,C16:0, C18:0, C24:1(15Z)について合成β-GlcCerを用いてMincleリガンド活性を検討したところ,C24:1(15Z)が最も強いレポーター活性を示した.疎水性側鎖に含まれる二重結合がMincleとの結合に何らかの影響を及ぼしていることが考えられるが,その理由は不明であり,Mincleとβ-GlcCerの共結晶構造解析の進展が待たれる.
6. β-GlcCerは免疫活性化能を発揮する細胞内代謝物である
次にβ-GlcCerとMincleの結合が,ミエロイド細胞を活性化しうるかを検討したところ,β-GlcCerは直接BMDCを活性化し炎症性サイトカイン,ケモカインの産生を誘導することが明らかとなった.加えてβ-GlcCerは,樹状細胞においてCD80/86, CD40といった共刺激分子やMHC class II分子の発現を上昇させた.これらの応答は,いずれもMincleを欠損させると抑制されたことから,Mincleはβ-GlcCerによるミエロイド細胞活性化において必須の受容体であることが示された.
Mincleは,自然免疫活性化を介して獲得免疫誘導に寄与するアジュバント受容体としても機能する9, 10).今回同定したβ-GlcCerも,獲得免疫の評価として卵白アルブミン(OVA)特異的T細胞応答をin vitro, in vivoについて検討したところ,どちらにおいても抗原特異的な獲得免疫応答を誘導しうることが明らかとなった.
β-GlcCerは,抗腫瘍効果を有することも報告されている30–32).この機序については,直接の細胞障害活性やNKT細胞の寄与等が提唱されてはいるが,詳細は不明である.Mincleを介した獲得免疫応答活性化も少なからず寄与しているのかもしれない.
7. 内因性β-GlcCerは組織損傷に伴う炎症を増強する
これまでの実験から,β-GlcCerは少なくとも,外から加えた際にMincleを介した免疫活性化能を発揮することが明らかとなった.しかし,β-GlcCerは内因性の糖脂質である.生理的条件下において内因性β-GlcCerは,何かしらの生理的機能を発揮するのだろうか.
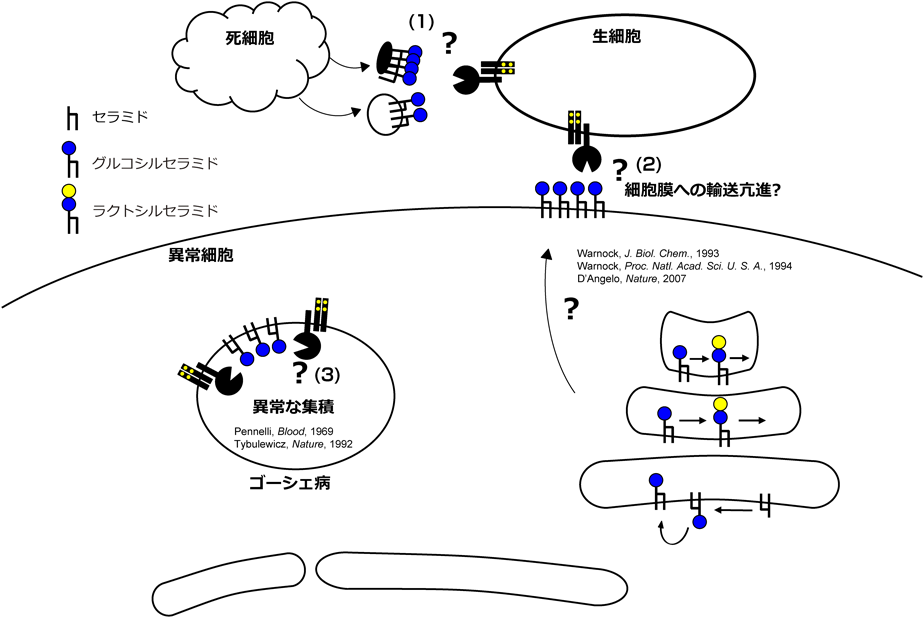
β-GlcCerは,その代謝制御が厳密に行われている.制御を担う酵素の一つである,β-GlcCer分解酵素(β-glucocerebrosidase:GBA1)の変異は,全身性にβ-GlcCer蓄積を引き起こしゴーシェ病の原因となる33).ゴーシェ病は,GBA1遺伝子の変異,または欠損により発症するリソソーム病の一種であり,肝脾腫,骨疾患,神経障害などの症状を特徴とする.β-GlcCer分解酵素補充療法が治療の第一選択であることから,β-GlcCerの蓄積がその病態形成に大きく関わっていることが考えられるが,発症機序は不明である.ゴーシェ病では,各組織や血漿中の免疫細胞の活性化がみられる.特に脳組織において過剰に起こるミクログリアの活性化が,神経機能障害に関わることが示唆され34, 35),この過剰活性化と病態発症の関連が注目されてはいるが,β-GlcCerの蓄積と免疫細胞活性化をつなぐ分子メカニズムの詳細はまったくわかっていなかった.
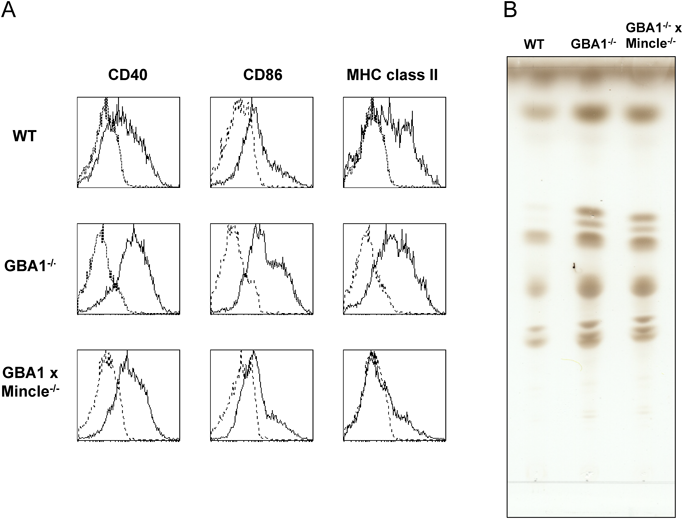
GBA1欠損マウスは出産直後致死となる36).このため,欠損マウス由来の胎仔肝細胞を野生型に移植することで,血球系細胞のみでGBA1を欠損するマウスを樹立した.このマウスでは,予想どおりβ-GlcCerの蓄積が観察されており,炎症応答に対する蓄積β-GlcCerの機能を解析したところ,放射線照射によって誘導される胸腺細胞死に付随して起こる胸腺への好中球浸潤が顕著に増強することが明らかとなった.この効果は,GBA1×Mincle二重欠損マウスでは抑制された(図5A, B).このとき,胸腺細胞死の度合いは各マウス間で同程度であったことから(図5C),細胞死に伴って放出されるβ-GlcCerが,Mincleを介して炎症惹起に作用することが示唆された.
神経系特異的にGBA1を欠損させたモデルマウスでは,早期からミクログリアの活性化と脳組織での炎症が認められ,これが神経障害を引き起こしていることが示唆されている34, 35).β-GlcCerの組成は各組織において異なることが知られている37)が,Mincleリガンド活性の強いC24:1(15Z)は,神経障害を伴う重篤なゴーシェ病患者の脳においても蓄積が観察される38).加えて,Mincleはミクログリアに発現が認められることから39),この機構にMincleが寄与している可能性も考えられる.Mincleのような活性化受容体による過剰炎症が,神経障害発症に寄与する新たな要因と考えられるかもしれない.
8. β-GlcCerは内因性アジュバントとして機能する
GBA1欠損樹状細胞は,β-GlcCerの蓄積に加え,培養すると外からの刺激がない条件下でも共刺激分子とMHC class IIの発現上昇が観察される.Mincleを欠損させるとこの発現上昇はみられなくなるが(図6A),同様にβ-GlcCerの蓄積が観察されることから(図6B),蓄積した内因性のβ-GlcCerが,Mincleを介して細胞の活性化に働くことが推測された.CD45.1陽性野生型樹状細胞と,CD45.2陽性GBA1欠損樹状細胞を共培養すると,CD45.1陽性野生型樹状細胞の活性化も認められ,GBA1欠損細胞だけでなく周囲の細胞も活性化することが示唆された.
蓄積β-GlcCerによってもたらされる活性化が,獲得免疫誘導に及ぼす影響を検討するため,GBA1欠損樹状細胞のT細胞活性化能を,BMDCとOVA特異的T細胞受容体を持つOT-IIトランスジェニックマウス由来のT細胞の共培養系を用いて検討した.GBA1欠損樹状細胞を用いたときには,野生型に比べて強力なT細胞応答がOVA抗原依存的に観察された.この反応はMincleを過剰発現させることでさらに増強し,欠損することで失われたことから,内因性β-GlcCerがMincleを介して,抗原特異的T細胞の効率的な活性化に寄与することが示唆された.OVAをパルスしたGBA1欠損樹状細胞をマウスに免疫した後の獲得免疫誘導の効率性をin vivoにて検討するため,免疫後のマウスから脾臓細胞を回収し,OVA抗原に対するリコール反応を検討した.GBA1欠損樹状細胞は,in vivoにおいても,野生型に比べて抗原特異的T細胞応答を有意に増強する結果が得られた.この増強効果もMincleの欠損により失われたため,β-GlcCerはMincleを介して内因性アジュバントとして機能すると考えられた.
ゴーシェ病モデルマウスでは,T細胞応答がTh1/Th17型に偏ることが報告されている40).Mincleは,アジュバント作用を担う受容体としてTh1/17応答を誘導することから9, 10),モデルマウスにおいてみられるこの現象は,蓄積したβ-GlcCerがMincleとの相互作用によって内因性アジュバントとして機能した結果かもしれない.実際,ゴーシェ病患者由来血液に含まれる単球上のCD1分子や,HLA-DR,-DP,-DQ分子は発現が上昇している41).これら抗原提示分子の発現上昇も,蓄積β-GlcCerがもたらす現象の一つとも考えられる.
9. β-GlcCer-Mincle経路とゴーシェ病
ゴーシェ病患者由来の血清中の炎症性サイトカイン,ケモカイン量を計測した報告では,M-CSFやIL-8の上昇が重症度(臓器肥大,血球減少)と相関していることが示唆されており42),蓄積β-GlcCerによるMincleを介した各ミエロイド細胞の活性化が推測される.
一方,GBA1欠損マウスの胎生致死は,GBA1とMincleを二重欠損することでは解消されなかった.GBA1欠損マウスの死亡原因は,主にセラミドの枯渇による皮膚形成の異常と,スフィンゴ糖脂質代謝異常による神経発生障害と考えられており36, 43),これらは免疫系の異常に伴う現象ではないと考えられる.今後,時期・組織特異的にGBA1を欠損させたり,変異GBA1を発現させる新たなモデルの樹立が,疾患における免疫系の寄与を検討する系として有効となってくるかもしれない.
近年,GBA1の阻害剤であるconduritol B epoxide(CBE)投与によって発症するゴーシェ病モデルマウス,また変異GBA1発現モデルにおいて,β-GlcCerに対する自己抗体が産生され,血清中に含まれる補体C5a量が上昇し,これが炎症増悪化に寄与している可能性が報告された44).また,このモデルにおいてC5aの受容体であるC5aR1を介するシグナルはβ-GlcCerのさらなる増加を誘導しており,著者らは,この経路の阻害が,ゴーシェ病の治療として有効である可能性を示唆している.ところが,β-GlcCerのような自己に豊富に存在する糖脂質に対してどのように抗体が産生されうるのか,C5a産生増加の機序は何か,C5aR1がどのようにしてβ-GlcCer産生を増加させるのか,など,免疫学的解析は不十分であり,今後の検証が待たれる.
ゴーシェ病は,パーキンソン病との関連が示唆されている45).このメカニズムとしては,蓄積したβ-GlcCerがα-シヌクレインの重合を誘導することで,神経毒性を有するアミロイドが形成されることが提唱されている46).しかし,パーキンソン病も炎症に伴い増悪が認められる疾患であることから,β-GlcCer-Mincle経路を介した過剰炎症の,パーキンソン病発症への寄与が予測される.Mincleが惹起する過剰炎症による神経障害への寄与が明らかとなることで,重篤な症状を呈する神経変性疾患に対する治療方法の幅が広がることを期待したい.
自己の異常は,普段と異なる局在,形態という形でさまざまな自然免疫受容体に伝えられる.CLRsのうち,DNGR-1(Clec9a)は局在の異常によって死細胞を認識する受容体である47).DNGR-1は,単量体のG(globular)-アクチンが重合したF(filamentous)-アクチンを認識することが報告されている48, 49).マウスでは,DNGR-1は交差提示(cross presentation)を行うCD8α+樹状細胞に主に発現しており,この認識が交差提示を介した細胞障害性T細胞の活性化に寄与していることが報告されている50, 51).DNGR-1とF-actinの結合様式は結晶構造解析から明らかとなっており,二つのF-actinがより合わさって形成される溝にDNGR-1は結合する52).F-actinは,アクチンフィラメント構造をとることで細胞の形状変化を担うタンパク質であり,通常細胞質に存在する.この分子は,細胞損傷によって細胞外に露出することでDNGR-1に認識される.このことは,DNGR-1がF-actinの認識を介して組織損傷を感知することを示唆している.
異常な形態の例としては,自己由来成分の結晶化があげられる.尿酸塩結晶(MSU)は尿酸血中濃度の上昇によって形成される結晶構造である.この分子は痛風の原因として知られているが,MICL(Clec12a)はこの結晶分子を認識する53).MICLは細胞内にITIM(immunoreceptor tyrosine-based inhibitory motif)を有し,リガンドが結合すると,活性化シグナルを担うタンパク質を脱リン酸化することで抑制性に働く.MSUは組織において炎症応答を引き起こすことから,MICLがこの応答を抑制すると推測される.しかし,MICLは痛風発症に関連の深いIL-1βの産生に影響を与えないことから,痛風発症時のMICLの働きは依然として不明のままである.さらに,ヒトMincleも結晶化した自己成分,コレステロール結晶を認識する2).コレステロール結晶の血管内皮への沈着は,炎症反応に伴い組織損傷を引き起こす.これが動脈硬化の原因の一つと考えられている.ヒトMincleを発現させたマウスを用いることで,このような種特異的な経路を評価することが可能になるかもしれない.
C型レクチン受容体がシグナルを誘導するためには,ある程度以上受容体が凝集することが必要である.そのためには何らかの形でvalencyを高める必要があり,上述の内因性リガンドはそれをよく説明する.β-GlcCerが生体内でリガンド活性を発揮するとき,どのようにしてこのvalencyを獲得しているのかは不明である.血清中の結合タンパク質の寄与,細胞外小胞膜としての放出,あるいは細胞形質膜への輸送54, 55)の亢進,あるいはオルガネラ膜上における膜ドメインの形成,など,さまざまな機構が考えられ(図7),今後の検討を要する.
本稿では,Mincleによる内因性糖脂質β-GlcCerの認識について紹介した.Mincleは,病原体のみならず,自己由来成分に対しても特定の糖脂質パターンを認識することで免疫応答を惹起する(図8).この応答は,感染防御のみならず生体恒常性に寄与する可能性が考えられるが,組織修復へのMincleの寄与に関する研究は不十分であり,今後の検討が必要である(図9).一方,病原体感染に伴い,細胞死が起こる例は多く知られている58, 59).感染局所において,病原体排除応答に内因性β-GlcCerが相加的,あるいは相乗的に寄与するような場面も考えられるかもしれない.通常は代謝中間体として働く糖脂質が,生体の危機を伝えるシグナルとなるという今回の知見は,細胞内に数多く存在する他の代謝産物も未知の機能を有することを想像させる.一方,これまで報告されているいわゆるDAMPs分子群の組織特異性,寄与率,冗長性,などの詳細はいまだ正確に解明されておらず,これら内因性免疫賦活物質を用いた人為的な免疫調節を実現していくためには,今後必要な課題であると考えられる.
謝辞Acknowledgments
本稿で紹介した研究は,九州大学生体防御医学研究所トランスオミクス医学研究センターメタボロミクス分野馬場健史先生,和泉自泰先生,九州大学薬学研究院臨床薬学部門宮本智文先生,国立感染症研究所細胞化学部第二室山地俊之先生との共同研究の成果であり,多大なご協力をいただきました.深く感謝の意を表します.
引用文献References
1) Mackenzie, K.J., Carroll, P., Martin, C.A., Murina, O., Fluteau, A., Simpson, D.J., Olova, N., Sutcliffe, H., Rainger, J.K., Leitch, A., Osborn, R.T., Wheeler, A.P., Nowotny, M., Gilbert, N., Chandra, T., Reijns, M.A.M., & Jackson, A.P. (2017) Nature, 548, 461–465.
2) Kiyotake, R., Oh-Hora, M., Ishikawa, E., Miyamoto, T., Ishibashi, T., & Yamasaki, S. (2015) J. Biol. Chem., 290, 25322–25332.
3) Miyake, K. & Kaisho, T. (2014) Curr. Opin. Immunol., 30, 85–90.
4) Roers, A., Hiller, B., & Hornung, V. (2016) Immunity, 44, 739–754.
5) Matsumoto, M., Tanaka, T., Kaisho, T., Sanjo, H., Copeland, N.G., Gilbert, D.J., Jenkins, N.A., & Akira, S. (1999) J. Immunol., 163, 5039–5048.
6) Yamasaki, S., Ishikawa, E., Sakuma, M., Hara, H., Ogata, K., & Saito, T. (2008) Nat. Immunol., 9, 1179–1188.
7) Ohtsuka, M., Arase, H., Takeuchi, A., Yamasaki, S., Shiina, R., Suenaga, T., Sakurai, D., Yokosuka, T., Arase, N., Iwashima, M., Kitamura, T., Moriya, H., & Saito, T. (2004) Proc. Natl. Acad. Sci. USA, 101, 8126–8131.
8) Ishikawa, E., Ishikawa, T., Morita, Y.S., Toyonaga, K., Yamada, H., Takeuchi, O., Kinoshita, T., Akira, S., Yoshikai, Y., & Yamasaki, S. (2009) J. Exp. Med., 206, 2879–2888.
9) Schoenen, H., Bodendorfer, B., Hitchens, K., Manzanero, S., Werninghaus, K., Nimmerjahn, F., Agger, E.M., Stenger, S., Andersen, P., Ruland, J., Brown, G.D., Wells, C., & Lang, R. (2010) J. Immunol., 184, 2756–2760.
10) Miyake, Y., Toyonaga, K., Mori, D., Kakuta, S., Hoshino, Y., Oyamada, A., Yamada, H., Ono, K., Suyama, M., Iwakura, Y., Yoshikai, Y., & Yamasaki, S. (2013) Immunity, 38, 1050–1062.
11) Brown, G.D. & Crocker, P.R. (2016) Microbiol. Spectr., 4, 1–26.
12) Feinberg, H., Jegouzo, S.A., Rowntree, T.J., Guan, Y., Brash, M.A., Taylor, M.E., Weis, W.I., & Drickamer, K. (2013) J. Biol. Chem., 288, 28457–28465.
13) Weis, W.I., Taylor, M.E., & Drickamer, K. (1998) Immunol. Rev., 163, 19–34.
14) Furukawa, A., Kamishikiryo, J., Mori, D., Toyonaga, K., Okabe, Y., Toji, A., Kanda, R., Miyake, Y., Ose, T., Yamasaki, S., & Maenaka, K. (2013) Proc. Natl. Acad. Sci. USA, 110, 17438–17443.
15) Jegouzo, S.A., Harding, E.C., Acton, O., Rex, M.J., Fadden, A.J., Taylor, M.E., & Drickamer, K. (2014) Glycobiology, 24, 1291–1300.
16) Ishikawa, T., Itoh, F., Yoshida, S., Saijo, S., Matsuzawa, T., Gonoi, T., Saito, T., Okawa, Y., Shibata, N., Miyamoto, T., & Yamasaki, S. (2013) Cell Host Microbe, 13, 477–488.
17) Behler-Janbeck, F., Takano, T., Maus, R., Stolper, J., Jonigk, D., Tort Tarres, M., Fuehner, T., Prasse, A., Welte, T., Timmer, M.S., Stocker, B.L., Nakanishi, Y., Miyamoto, T., Yamasaki, S., & Maus, U.A. (2016) PLoS Pathog., 12, e1006038.
18) Stocker, B.L., Khan, A.A., Chee, S.H., Kamena, F., & Timmer, M.S. (2014) ChemBioChem, 15, 382–388.
19) van der Peet, P.L., Gunawan, C., Torigoe, S., Yamasaki, S., & Williams, S.J. (2015) Chem. Commun. (Camb.), 51, 5100–5103.
20) Decout, A., Silva-Gomes, S., Drocourt, D., Barbe, S., Andre, I., Cueto, F.J., Lioux, T., Sancho, D., Perouzel, E., Vercellone, A., Prandi, J., Gilleron, M., Tiraby, G., & Nigou, J. (2017) Proc. Natl. Acad. Sci. USA, 114, 2675–2680.
21) Wishart, D.S., Jewison, T., Guo, A.C., Wilson, M., Knox, C., Liu, Y., Djoumbou, Y., Mandal, R., Aziat, F., Dong, E., Bouatra, S., Sinelnikov, I., Arndt, D., Xia, J., Liu, P., Yallou, F., Bjorndahl, T., Perez-Pineiro, R., Eisner, R., Allen, F., Neveu, V., Greiner, R., & Scalbert, A. (2013) Nucleic Acids Res., 41(D1), D801–D807.
22) Nagata, M., Izumi, Y., Ishikawa, E., Kiyotake, R., Doi, R., Iwai, S., Omahdi, Z., Yamaji, T., Miyamoto, T., Bamba, T., & Yamasaki, S. (2017) Proc. Natl. Acad. Sci. USA, 114, E3285–E3294.
23) Yamaji, T. & Hanada, K. (2015) Traffic, 16, 101–122.
24) Brennan, P.J., Tatituri, R.V., Brigl, M., Kim, E.Y., Tuli, A., Sanderson, J.P., Gadola, S.D., Hsu, F.F., Besra, G.S., & Brenner, M.B. (2011) Nat. Immunol., 12, 1202–1211.
25) Kain, L., Webb, B., Anderson, B.L., Deng, S., Holt, M., Costanzo, A., Zhao, M., Self, K., Teyton, A., Everett, C., Kronenberg, M., Zajonc, D.M., Bendelac, A., Savage, P.B., & Teyton, L. (2014) Immunity, 41, 543–554.
26) Brennan, P.J., Tatituri, R.V., Heiss, C., Watts, G.F., Hsu, F.F., Veerapen, N., Cox, L.R., Azadi, P., Besra, G.S., & Brenner, M.B. (2014) Proc. Natl. Acad. Sci. USA, 111, 13433–13438.
27) Brennan, P.J., Cheng, T.Y., Pellicci, D.G., Watts, G.F.M., Veerapen, N., Young, D.C., Rossjohn, J., Besra, G.S., Godfrey, D.I., Brenner, M.B., & Moody, D.B. (2017) Proc. Natl. Acad. Sci. USA, 114, 8348–8353.
28) Izawa, K., Yamanishi, Y., Maehara, A., Takahashi, M., Isobe, M., Ito, S., Kaitani, A., Matsukawa, T., Matsuoka, T., Nakahara, F., Oki, T., Kiyonari, H., Abe, T., Okumura, K., Kitamura, T., & Kitaura, J. (2012) Immunity, 37, 827–839.
29) Patel, O., Pellicci, D.G., Gras, S., Sandoval-Romero, M.L., Uldrich, A.P., Mallevaey, T., Clarke, A.J., Le Nours, J., Theodossis, A., Cardell, S.L., Gapin, L., Godfrey, D.I., & Rossjohn, J. (2012) Nat. Immunol., 13, 857–863.
30) Symolon, H., Schmelz, E.M., Dillehay, D.L., & Merrill, A.H. Jr. (2004) J. Nutr., 134, 1157–1161.
31) Oku, H., Li, C., Shimatani, M., Iwasaki, H., Toda, T., Okabe, T., & Watanabe, H. (2009) Cancer Chemother. Pharmacol., 64, 485–496.
32) Inafuku, M., Li, C., Kanda, Y., Kawamura, T., Takeda, K., Oku, H., & Watanabe, H. (2012) Lipids, 47, 581–591.
33) Hruska, K.S., LaMarca, M.E., Scott, C.R., & Sidransky, E. (2008) Hum. Mutat., 29, 567–583.
34) Vitner, E.B., Farfel-Becker, T., Eilam, R., Biton, I., & Futerman, A.H. (2012) Brain, 135, 1724–1735.
35) Vitner, E.B., Farfel-Becker, T., Ferreira, N.S., Leshkowitz, D., Sharma, P., Lang, K.S., & Futerman, A.H. (2016) J. Neuroinflammation, 13, 104.
36) Tybulewicz, V.L., Tremblay, M.L., LaMarca, M.E., Willemsen, R., Stubblefield, B.K., Winfield, S., Zablocka, B., Sidransky, E., Martin, B.M., Huang, S.P., Mintzer, K.A., Westphal, H., Mulligan, R.C., & Ginns, E.I. (1992) Nature, 357, 407–410.
37) Ishibashi, Y., Kohyama-Koganeya, A., & Hirabayashi, Y. (2013) Biochim. Biophys. Acta, 1831, 1475–1485.
38) Burrow, T.A., Sun, Y., Prada, C.E., Bailey, L., Zhang, W., Brewer, A., Wu, S.W., Setchell, K.D., Witte, D., Cohen, M.B., & Grabowski, G.A. (2015) Mol. Genet. Metab., 114, 233–241.
39) McKimmie, C.S., Roy, D., Forster, T., & Fazakerley, J.K. (2006) J. Neuroimmunol., 175, 128–141.
40) Pandey, M.K., Rani, R., Zhang, W., Setchell, K., & Grabowski, G.A. (2012) Mol. Genet. Metab., 106, 310–322.
41) Balreira, A., Lacerda, L., Miranda, C.S., & Arosa, F.A. (2005) Br. J. Haematol., 129, 667–676.
42) Hollak, C.E., Evers, L., Aerts, J.M., & van Oers, M.H. (1997) Blood Cells Mol. Dis., 23, 201–212.
43) Sidransky, E., Sherer, D.M., & Ginns, E.I. (1992) Pediatr. Res., 32, 494–498.
44) Pandey, M.K., Burrow, T.A., Rani, R., Martin, L.J., Witte, D., Setchell, K.D., McKay, M.A., Magnusen, A.F., Zhang, W., Liou, B., Kohl, J., & Grabowski, G.A. (2017) Nature, 543, 108–112.
45) Aharon-Peretz, J., Rosenbaum, H., & Gershoni-Baruch, R. (2004) N. Engl. J. Med., 351, 1972–1977.
46) Mazzulli, J.R., Xu, Y.H., Sun, Y., Knight, A.L., McLean, P.J., Caldwell, G.A., Sidransky, E., Grabowski, G.A., & Krainc, D. (2011) Cell, 146, 37–52.
47) Sancho, D., Joffre, O.P., Keller, A.M., Rogers, N.C., Martinez, D., Hernanz-Falcon, P., Rosewell, I., & Reis e Sousa, C. (2009) Nature, 458, 899–903.
48) Ahrens, S., Zelenay, S., Sancho, D., Hanc, P., Kjaer, S., Feest, C., Fletcher, G., Durkin, C., Postigo, A., Skehel, M., Batista, F., Thompson, B., Way, M., Reis e Sousa, C., & Schulz, O. (2012) Immunity, 36, 635–645.
49) Zhang, J.G., Czabotar, P.E., Policheni, A.N., Caminschi, I., Wan, S.S., Kitsoulis, S., Tullett, K.M., Robin, A.Y., Brammananth, R., van Delft, M.F., Lu, J., O’Reilly, L.A., Josefsson, E.C., Kile, B.T., Chin, W.J., Mintern, J.D., Olshina, M.A., Wong, W., Baum, J., Wright, M.D., Huang, D.C., Mohandas, N., Coppel, R.L., Colman, P.M., Nicola, N.A., Shortman, K., & Lahoud, M.H. (2012) Immunity, 36, 646–657.
50) Zelenay, S., Keller, A.M., Whitney, P.G., Schraml, B.U., Deddouche, S., Rogers, N.C., Schulz, O., Sancho, D., & Reis e Sousa, C. (2012) J. Clin. Invest., 122, 1615–1627.
51) Iborra, S., Izquierdo, H.M., Martinez-Lopez, M., Blanco-Menendez, N., Reis e Sousa, C., & Sancho, D. (2012) J. Clin. Invest., 122, 1628–1643.
52) Hanc, P., Fujii, T., Iborra, S., Yamada, Y., Huotari, J., Schulz, O., Ahrens, S., Kjaer, S., Way, M., Sancho, D., Namba, K., & Reis, E.S.C. (2015) Immunity, 42, 839–849.
53) Neumann, K., Castineiras-Vilarino, M., Hockendorf, U., Hannesschlager, N., Lemeer, S., Kupka, D., Meyermann, S., Lech, M., Anders, H.J., Kuster, B., Busch, D.H., Gewies, A., Naumann, R., Gross, O., & Ruland, J. (2014) Immunity, 40, 389–399.
54) D’Angelo, G., Polishchuk, E., Di Tullio, G., Santoro, M., Di Campli, A., Godi, A., West, G., Bielawski, J., Chuang, C.C., van der Spoel, A.C., Platt, F.M., Hannun, Y.A., Polishchuk, R., Mattjus, P., & De Matteis, M.A. (2007) Nature, 449, 62–67.
55) Warnock, D.E., Roberts, C., Lutz, M.S., Blackburn, W.A., Young, W.W. Jr., & Baenziger, J.U. (1993) J. Biol. Chem., 268, 10145–10153.
56) Warnock, D.E., Lutz, M.S., Blackburn, W.A., Young, W.W. Jr., & Baenziger, J.U. (1994) Proc. Natl. Acad. Sci. USA, 91, 2708–2712.
57) Pennelli, N., Scaravilli, F., & Zacchello, F. (1969) Blood, 34, 331–347.
58) Weinrauch, Y. & Zychlinsky, A. (1999) Annu. Rev. Microbiol., 53, 155–187.
59) Jorgensen, I., Rayamajhi, M., & Miao, E.A. (2017) Nat. Rev. Immunol., 17, 151–164.
著者紹介Author Profile
 永田 雅大(ながた まさひろ)
永田 雅大(ながた まさひろ)大阪大学微生物病研究所分子免疫制御分野特任研究員.博士(医学).
略歴2011年九州大学医学部生命科学科卒業.13年同大学院医学系学府医科学専攻修士課程修了.17年同大学院医学系学府医学専攻博士課程単位修得退学を経て,同年同大学院にて博士号取得.同年より現職.
研究テーマと抱負C型レクチン受容体による自己・非自己認識機構について研究を行っております.近年では,自己,非自己に関わらず,生体にとって危険な状況を特定の分子パターンが示していることが示唆されており,その状況を免疫系がどのように厳密に識別しているのか明らかにしたいと考えています.
趣味サッカー,フットサル.