がんが原発巣から転移して体内で拡散した場合,予後が悪くなる.一方,がんが転移せずに局所で増殖する場合では切除治療が可能であり,比較的予後がよい.前者のがんは一般に播種型,後者は圧排性増殖型のがん浸潤を原因とする.二つのがん浸潤モードの選択メカニズムは明らかではない.
近年,播種型のがん浸潤として,がん細胞と間質細胞からなるヘテロな細胞集団の浸潤システムが注目されている.多くのがん細胞株は近接する間質の線維芽細胞に作用し,がん関連線維芽細胞(cancer associated fibroblast:CAF)へと変貌させる1).CAFはがん細胞に先立って組織深部へと浸潤するようになり,がん細胞の浸潤をより広域へとリードすることがわかってきた.また,がん細胞とCAFとの相互作用はがんの増殖や転移に適したがん微小環境の整備にも関与する2).このように,間質層でのがん悪性化はCAFにより飛躍的に亢進することが考えられている.がん微小環境の形成は細胞集団間におけるさまざまなサイトカイン,ケモカイン,細胞外マトリックス,細胞外小胞の産生および応答により促進することが報告されている3).
一方で,間質細胞はがんに対する抑制的役割も担っている.線維芽細胞は,いくつかのがん細胞株のアポトーシスを誘発する.たとえば,間葉系幹細胞ではTRAILを発現し,TRAIL感受性であるがん細胞のアポトーシスを引き起こす.このように間質細胞はがんに対して相反する影響を担っている4).
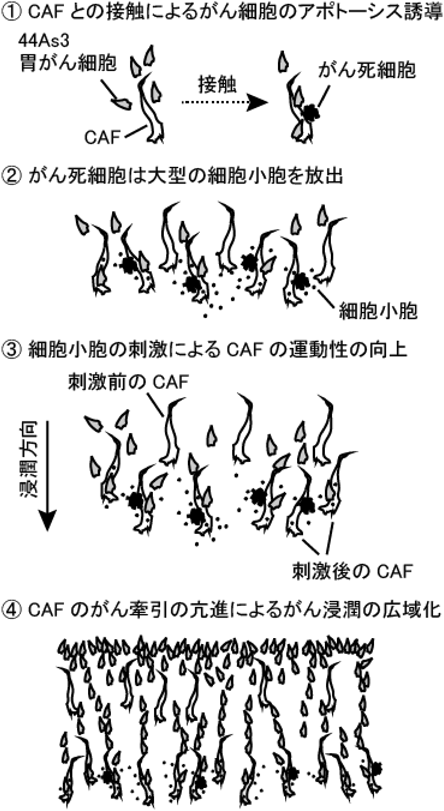
著者らは,がん細胞と間質細胞の細胞間応答について調べてきた.最近,播種型と圧排性増殖型のがん浸潤モードの選択につながる新規の浸潤システムを報告している5).まず,CAFによるスキルス胃がん細胞のアポトーシス誘導を見つけた.CAFと接触したがん細胞は大型の細胞外小胞(Apo-EVs)を放出し,これに刺激されたCAFは運動性を向上させることがわかった(図1).
このがん細胞死はがん浸潤を大きく変動させた.胃がん細胞とCAFをゲル上で共培養した場合,Apo-EVsに刺激されたCAFはゲル深部へと浸潤し,引き続きがん細胞を牽引することで広範囲の播種型がん浸潤となった.一転して,アポトーシス阻害剤ZVADの添加によりがんアポトーシスを抑制するとCAFの浸潤は減衰し,がん細胞は圧排性増殖型のゲル浅部での浸潤を増加させた.さらに,これらがん浸潤システムがマウス個体でも反映されることを示した.これまでに,がん細胞のアポトーシスは腫瘍関連マクロファージとの相互作用によりがん増殖を亢進する働きがあることが報告されている6).著者らが明らかにしたがん浸潤システムはがんアポトーシスを原因としたがん悪性化につながる新たなメカニズムといえる.
CAFとがん細胞はさまざまなサイトカイン,ケモカイン,細胞外マトリックスの分泌により互いを制御する.エクソソームは細胞より細胞外空間へと分泌される30~100 nmの細胞外小胞であるが,近年,エクソソームは隣接または遠隔の細胞間クロストークに重要であることがわかってきた.エクソソームはシグナルペプチド,microRNA(miRNA),脂質,DNAなどの多様な生物活性因子を含むため,多岐にわたるがんシグナル伝達に関与することがわかってきた.腫瘍細胞では多量のエクソソームが分泌され,がん微小環境をより広域とさせる作用をもたらす.
エクソソームmiRNAは安定的に運搬されることが知られており,遠隔の細胞でも取り込まれ,特定の遺伝子発現を調節している.エクソソームは内包の多種のmiRNAによって,発がん過程のがん増殖,浸潤,がん幹細胞増殖など腫瘍進展にとって有利ながん微小環境を作り出せることが明らかとなってきた.
エクソソームmiRNAはCAF,正常線維芽細胞(normal fibroblast:NF),およびがん細胞の細胞間クロストークに重要である.たとえば,miR-9は乳がんの腫瘍細胞からのエクソソームによりNFへと運搬され,CAF様表現型を誘導する.miR-9はNFからも放出され,腫瘍細胞へと運ばれて細胞移動を促進する7).また,miR-409の過剰発現でもNFのCAF様表現型の誘導は認められ,このNFから分泌されるエクソソームmiR-409は腫瘍の誘導および前立腺がん細胞のEMT(上皮間葉転換:上皮細胞としての特性を失い周辺組織に移動しやすい間葉系細胞としての特徴を獲得する現象)を促進することが報告されている8).
miR-451は腫瘍抑制因子としての一面を有し,さまざまな腫瘍型で減少している.最近の研究では逆に,CAF由来のエクソソームmiR-451は腫瘍細胞の移動およびがん進行を促進する分子シグナルとして働くことがわかった.したがってmiRNAはがん細胞,CAF,およびエクソソームにおいて異なる役割があるかもしれない9).
ヒト間葉系幹細胞(mesenchymal stem/stromal cell:hMSC)はサイトカインTNF-αにより活性化した場合,腫瘍壊死因子関連アポトーシス誘導リガンド(tumor necrosis factor-related apoptosis-inducing ligand:TRAIL)の発現を上昇させ,in vitroおよびin vivoでのMDA(MDA-MB-231)乳がん細胞のアポトーシスを誘導する.また,活性化hMSCはMDA細胞のアポトーシスを誘導するだけでなく,MDA細胞の転移性を減少させる.活性化hMSCと反応させたMDA細胞が乳腺脂肪体に埋め込まれた場合,腫瘍性が低下し,肺転移を抑制することが明らかとなった.さらに,活性化hMSCと反応させたMDA細胞でもTRAILの発現が増加することが示された.MDA細胞におけるTRAILの高発現は,活性化hMSCから分泌されるインターフェロンβ(IFN-β)による刺激が原因であると考えられた.さらに,活性化hMSCでのIFN-β産生はMDAアポトーシス細胞から放出されたRNAおよびDNAによって誘導されていた.
また,乳がん患者から単離されたCAFでも同様にDNAおよびRNA刺激時にTRAILおよびIFN-βを発現することがわかった.このことからもTRAIL感受性がん細胞と間質細胞のクロストークによる腫瘍抑制効果を標的としたTRAIL感受性がんの新規治療法の開発が期待できる.
4. 胃がん細胞のアポトーシスはCAFとの接触により誘導される
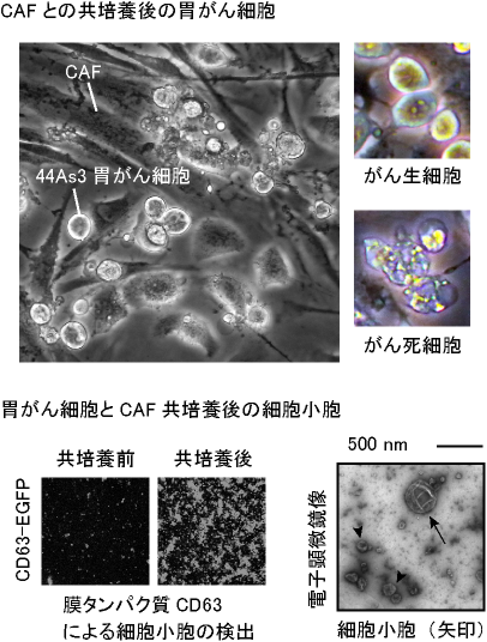
著者らはCAFと44As3スキルス胃がん細胞を共培養した際,がん細胞がアポトーシス様の形態で死ぬことを見いだした(図2).共培養後,アポトーシスの特徴である核の凝集や断片化が25%の44As3細胞で観察されたが,CAFでは観察されなかった.このがんアポトーシス誘導においては,CAFと44As3細胞との接触が重要であった.共培養後,両細胞間での頻繁な接触が観察され,アポトーシス44As3細胞はCAFの表面上に付着していた.一方,CAFの培養上清を回収し,この上清により44As3細胞を培養した場合ではアポトーシスは低頻度となった.
5. DR4はCAF誘導性のがんアポトーシスに関連する
44As3死細胞はTUNEL陽性であり,活性型のカスパーゼ3と8に対しても陽性であった.また,全カスパーゼ阻害剤ZVADで処理後,CAF誘導性のがんアポトーシスは抑制され,cleaved caspase-3(活性型カスパーゼ3)が減衰した.
CAFと胃がん細胞株でのアポトーシス関連分子の発現を調べた結果,デス受容体DR4(death receptor-4),DR4と物理的に関連するアダプター受容体FADDおよびカスパーゼ8が胃がん細胞で高発現していた.これらはCAFでは低発現であった.同様な発現はin vitro/in vivoで確認された.DR4発現をsiRNAによりノックダウンした44As3細胞では,一連のがんアポトーシスを部分的に抑制できた.また,DR4低発現であるHSC-59胃がん細胞ではCAF誘導性のアポトーシスを受けない.これらはDR4の媒介するアポトーシス経路とがん細胞死の関連を示唆している.
6. CAF誘導性のがんアポトーシスは,がん細胞の浸潤モードを転換させる
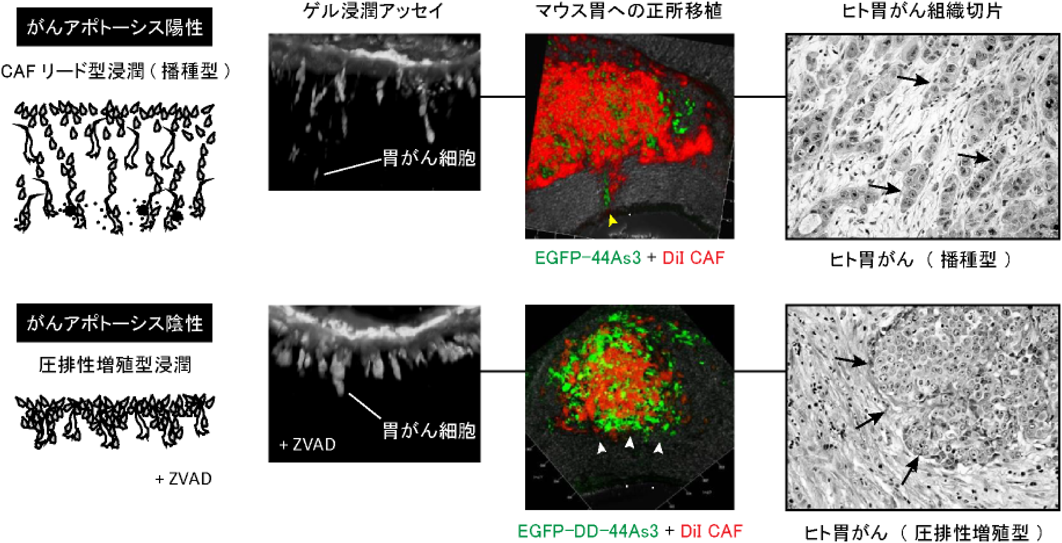
そこで,CAF誘導性のがんアポトーシスとがん浸潤との関連を検討するため,44As3細胞とCAFを細胞外マトリックス含有ゲル上で培養し,両細胞の浸潤過程を詳細に解析した(図3,ゲル浸潤アッセイ).
著者らは2種のがん浸潤モードをこのアッセイにより見いだした.一つは,CAFががん細胞に先立ってゲル深部へと浸潤し,がん細胞がCAFを追従して深部・広域へと進行するCAFリード型のがん浸潤(播種型に相当),もう一つは44As3細胞がクラスターを形成し,ゲル浅部で進行する圧排性増殖型に近い浸潤である.ちなみに正常線維芽細胞との共培養ではがん浸潤は認められない.
次に,浸潤過程の44As3細胞のアポトーシスを調べた.ヒストンH2B-EGFP-44As3細胞により,特にCAFの密度の高い領域において核の凝縮または断片化が認められた.ZVAD添加により,このアポトーシスは抑制された.ZVAD処理後のCAFと44As3細胞の浸潤を観察すると,CAFのゲル内への浸潤距離が短くなり,CAFリード型のゲル深部へのがん浸潤は減少した.代わりに圧排性のがん浸潤が広範な領域を占めることがわかった.以上の結果から,CAF誘導性のがん細胞アポトーシスはがん浸潤モード転換の鍵であることが示唆された.
7. アポトーシス44As3細胞から放出された細胞外小胞はCAFの浸潤を促進する
CAFの浸潤はがんアポトーシスの影響を受けたことから,がん死細胞の産生する細胞外小胞(EV)によるCAF浸潤性亢進の可能性を検証した.
EVマーカーであるCD63-EGFPを発現する44As3細胞とCAFを共培養した培地からEVを収集し,ZVAD処理の有無によるEVの性質およびCAF活性化への影響を調べた.ZVAD処理後にがんアポトーシスは抑制され,EV総量は減少した.また,蛍光・電子顕微鏡によりEV口径を比較すると,アポトーシス細胞では小型なEVであるエクソソーム・マイクロベシクルとともに大型なEV(Apo-EVs)産生(>200 nm)を示した.
CAFは本質的に高度な浸潤性を備えているが,得られたEVの添加はその浸潤性を亢進することがわかった.一方,ZVAD処理後に収集したEVはCAF浸潤を促進しない.また,遠心分離を用いてEVsを分画・収集し,Apo-EVsとエクソソームを得た.Apo-EVsは線維芽細胞の浸潤性を亢進させた.このようにがん死細胞の放出する特異的EVはCAFを刺激し,CAFリード型がん浸潤を亢進させていた.
8. デスドメイン(DD)フラグメントの発現はCAF誘導のがんアポトーシスを抑制する
著者らはCAF誘導性アポトーシスの恒常的な抑制を目指した.CAF誘導性のがんアポトーシスは部分的にDR4-カスパーゼ8シグナル伝達により媒介されたので,アポトーシス誘導はDRおよびプロカスパーゼ8, FADDで形成されるデス複合体を介して進むと考えた10).FADDのdeath domain(以下DD)フラグメントはデス複合体形成を妨害するドミナントネガティブとしての作用が報告されている11).そこで,EGFP-DDを44As3細胞に発現させ(EGFP-DD-44As3細胞),マウス個体でのがんアポトーシスを抑制できるか検討した.CAFおよびEGFP-44As3またはEGFP-DD-44As3細胞をヌードマウスに皮下移植した.TUNELアッセイにより腫瘍部のアポトーシスを調べた結果,EGFP-DD-44As3細胞のアポトーシスはEGFP-44As細胞を移植した場合よりも有意に低くなった.
9. がんアポトーシスは腫瘍を悪性化させる可能性がある
著者らは,in vitroのゲル浸潤アッセイによりCAFとEGFP-DD-44As3細胞をゲル上にて共培養した場合,CAFリード型のがん浸潤は抑制され,ZVAD処理時に観察される圧排性増殖型のがん浸潤が増加することを明らかにした.
次に,生体でのがんアポトーシスとがん浸潤の関連を調べるため,CAFとともにEGFP-44As3(コントロール)またはEGFP-DD-44As3細胞をヌードマウスの胃壁に移植した.粘膜下層に分布した44As3細胞とCAFの浸潤過程を追跡した結果,EGFP-44As3細胞とCAF移植5日後に,CAFががん浸潤に先立って筋肉層に侵入するCAFリード型のがん浸潤が認められた.一方,EGFP-DD-44As3細胞とCAFを移植した場合では,両方の細胞は筋層の表面領域のみに存在し,筋層内には入らなかった.移植16日後,EGFP-44As3細胞により形成される腫瘍は不定形となり小さな結節の播種が確認されたが,EGFP-DD-44As3細胞の形成する腫瘍では胃壁における圧排性増殖型のがん浸潤に収まった.また,EGFP-44As3細胞のリンパ節転移が認められたが,EGFP-DD-44As3細胞の移植では観察されなかった.
以上の結果から,CAF誘導のがんアポトーシスが生存したがん細胞のCAFリード型がん浸潤を促進し,組織深部や他臓器へのがんの播種・転移を導くことが示唆された.
NFもCAFも一定量のがん細胞を殺す.がん浸潤を促進しないNFではがん抑制となる作用が,一転してCAFではがん組織の浸潤様式を転換することでむしろがんの進展を促すことがわかった.現在,がん死細胞のEVに誘導されるCAF活性化シグナルとしてmiRNAに注目して解明を進めている.また,CAF誘導性のがんデスシグナルの詳細も調べている.CAFを不活性とさせ,がんアポトーシス誘導に働く一面のみを亢進できれば効果的ながん治療につながることが期待される.
引用文献References
1) Hwang, R.F., Moore, T., Arumugam, T., Ramachandran, V., Amos, K.D., Rivera, A., Ji, B., Evans, D.B., & Logsdon, C.D. (2008) Cancer Res., 68, 918–926.
2) Hsu, H.S., Lin, J.H., Hsu, T.W., Su, K., Wang, C.W., Yang, K.Y., Chiou, S.H., & Hung, S.C. (2012) Lung Cancer, 75, 167–177.
3) Yang, F., Ning, Z., Ma, L., Liu, W., Shao, C., Shu, Y., & Shen, H. (2017) Mol. Cancer, 16, 148.
4) Yoon, N., Park, M.S., Shigemoto, T., Peltier, G., & Lee, R.H. (2016) Cell Death Dis., 7, e2191.
5) Itoh, G., Chida, S., Yanagihara, K., Yashiro, M., Aiba, N., & Tanaka, M. (2017) Oncogene, 36, 4434–4444.
6) Lauber, K. & Herrmann, M. (2015) Curr. Biol., 25, 198–201.
7) Baroni, S., Romero-Cordoba, S., Plantamura, I., Dugo, M., D’Ippolito, E., Cataldo, A., Cosentino, G., Angeloni, V., Rossini, A., Daidone, M.G., & Iorio, M.V. (2016) Cell Death Dis., 7, e2312.
8) Josson, S., Gururajan, M., Sung, S.Y., Hu, P., Shao, C., Zhau, H.E., Liu, C., Lichterman, J., Duan, P., Li, Q., Rogatko, A., Posadas, E.M., Haga, C.L., & Chung, L.W. (2015) Oncogene, 34, 2690–2699.
9) Khazaei, S., Nouraee, N., Moradi, A., & Mowla, S.J. (2017) Clin. Transl. Oncol., 19, 633–640.
10) Lo, Y.C., Lin, S.C., Yang, C.Y., & Tung, J.Y. (2015) Apoptosis, 20, 124–135.
11) Sakamaki, K., Takagi, C., Kominami, K., Sakata, S., Yaoita, Y., Kubota, H.Y., Nozaki, M., Yonehara, S., & Ueno, N. (2004) Genes Cells, 9, 1249–1264.
著者紹介Author Profile
 伊藤 剛(いとう ごう)
伊藤 剛(いとう ごう)秋田大学大学院医学系研究科医学専攻分子生化学講座助教.Ph.D.
略歴1978年大分県出身.2002年山口大学卒業,08年同大学院理工学研究科(祐村恵彦教授)修了.東北大学加齢医学研究所分子腫瘍学研究分野(田中耕三教授)にて08~11年博士研究員,11~14年助教.14年より現職.
研究テーマと抱負間質細胞と協調した癌浸潤.分裂期の染色体分配および細胞質分裂メカニズム.癌アポトーシス.細胞性粘菌.哺乳類培養細胞.「抱負は良い研究をコスパ良く,数理解析のスキルアップ」.
ウェブサイトhttp://www.med.akita-u.ac.jp/~seika2/
趣味ブックオフ・ハードオフ巡り.
田中 正光(たなか まさみつ)秋田大学大学院医学系研究科医学専攻分子生化学講座教授.博士(医学).
略歴1962年静岡県に生る.87年浜松医科大学卒業.92年同大学院博士課程修了.94年ハーバード大学医学部ポスドク.96年浜松医科大学病理学第一講座助教.2003年国立がん研究センター研究所室長.09年より現職.
研究テーマと抱負がんの進展に伴う間質細胞の動態を調べる事で,がん組織の情報がいかに周辺組織に伝達され拡散してゆくのか明らかにし,腫瘍形態の病理像にフィードバックしてゆきたい.
ウェブサイトhttp://www.med.akita-u.ac.jp/~seika2/